En este capítulo se presenta una información que permitirá al estudiante conocer los métodos de análisis utilizados en el examen técnico del hierro y del acero. La naturaleza de este trabajo impide un tratamiento detallado del tema -un tratamiento que puede exigir una estimación de una o más de las siguientes sustancias:-Carbono (libre y combinado), azufre, silicio, fósforo, manganeso, titanio, cobre, níquel, cobalto, cromo, aluminio, arsénico, antimonio, estaño, tungsteno, vanadio, nitrógeno, hierro. En general, las estimaciones más necesarias son las del carbono, el azufre, el silicio y el fósforo. De los otros elementos y compuestos mencionados, se requieren determinaciones de uno o más en el caso de los aceros especiales. Para obtener información sobre estas determinaciones se remite al estudiante a Chemical Analysis and Foundry Chemistry, de Crobaugh; The. Chemical Analysis of Iron, de Blair; «Carbon in Steel by Direct Combustion», de Blount, en The Analyst, enero de 1902; «Sulphur in Wrought Iron and Steel», de Auchy, en el Jour. Amer. Chem. Soc., marzo de 1901, y otros artículos en las mismas revistas. El estudiante que desee ir más allá debe, si es posible, obtener acceso a los trabajos y artículos de Campbell, Drown y otros, publicados de vez en cuando en las diversas revistas químicas y metalúrgicas.
Como el tiempo del estudiante es limitado, puede posponer por el momento la estimación del silicio y del fósforo, aunque se dan por su importancia tanto para el metalúrgico como para el fundidor.
Para que el estudiante pueda obtener una comprensión más completa del tema, no estarán de más algunas notas sobre la composición y propiedades de las sustancias consideradas. En cuanto a la influencia de los distintos elementos en el acero, consúltese The Manufacture and Properties of Structural Steel, de H. H. Campbell.
El carbono existe en el hierro en tres estados: grafítico, disuelto y combinado. Además de éstas, se han identificado otras formas mediante el microscopio.
El azufre existe en el hierro principalmente como el sulfuro FeS. que es soluble en el hierro fundido.
El fósforo existe como fosfuro de hierro, que es completamente soluble en el hierro fundido.
El silicio forma siliciuro de hierro, que también es soluble en el hierro fundido.
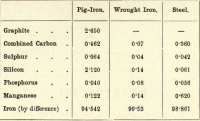
De estos cuatro elementos, pues, el carbono es el único que puede existir en estado libre. Las variaciones en las proporciones de los diversos elementos presentes son casi infinitas, pero la siguiente breve tabulación da la composición aproximada del arrabio, el hierro forjado y el acero, aunque cada uno de ellos está sujeto a una variación considerable.
El estudiante debe estimar lo siguiente:-
(1) Carbono,
(a) Total.
(b) Grafítico.
(c) Combinado.
(2) Azufre.
(3) Silicio. (Si el tiempo lo permite.)
(4) Fósforo. (Si el tiempo lo permite.)
CARBONO Total
En esta estimación el carbono se convierte en CO2 que se absorbe en potasa cáustica. A partir del peso del CO2 así obtenido se calcula el carbono.
A primera vista parecería que el procedimiento más sencillo sería encender los barrenos de hierro o acero directamente en una corriente de oxígeno y absorber el CO2 así formado en KHO. Desgraciadamente, este método, hasta ahora, ha resultado inexacto, o bien, cuando se obtenía una combustión completa, el aparato necesario para soportar la alta temperatura u otras variaciones de tratamiento no era adecuado para el trabajo técnico (véanse los artículos de Blount en The Analyst). El estudiante se dará cuenta de que el método que aquí se da no es en absoluto ideal, desde un punto de vista técnico, en lo que respecta a la comodidad y la rapidez, y parece probable que sea sustituido en un futuro próximo por algún método de oxidación directa más rápido.
Método adoptado.-Al consultar las numerosas obras sobre este tema, se encontrará una gran variedad de métodos. El método que aquí se presenta, con el cuidado habitual, dará resultados precisos. Brevemente es como sigue:-
El hierro se disuelve en una solución de doble cloruro de potasio y cobre, hecha ácida con HCl. El cobre metálico se precipita y se vuelve a disolver; el hierro se disuelve, quedando el carbón en suspensión. A continuación se recoge y se enciende en el horno de combustión con oxígeno, y se pesa el CO2 desprendido.
Solución del hierro.-Se pesa 1 gramo de borras de arrabio. Pasar a un vaso de precipitados de 300 c.c. Añadir 100 c.cs. Solución de CuCl2,2KCl,2H2O, que se hace como sigue. Disolver en agua 149,1 partes de KCl y 170,3 partes de CuCl2, 2H2O cristalizado. Evaporar y cristalizar el cloruro doble. Disolver 300 gms. de la sal doble en agua destilada. Filtrar a través de amianto encendido, y conservar en frascos de vidrio con tapón.
Al contenido del vaso de precipitados añadir 7 c.cs. HCl para que la solución sea ácida. Remover de forma intermitente hasta que se produzca la disolución del hierro. Colocar el vaso de precipitados y el contenido hacia el final de la solución en un baño de agua a una temperatura de unos 60° C. Se producen las siguientes reacciones: Fe + CuCl2 = FeCl2 + Cu y Cu + CuCl2 = 2CuCl. El KCl simplemente ayuda a la solución del cobre precipitado. En unos 40 minutos desde la adición de los cloruros dobles, la solución debería estar casi completa y la mayor parte del cobre disuelto. Lavar las paredes del vaso de precipitados con un poco de doble cloruro acidulado. A la solución se le añade un poco de amianto encendido para sedimentar la materia carbonosa y evitar que obstruya el filtro (como recomienda Barba).
Para la filtración, son muy convenientes los botes especiales de platino, instalados según el principio del crisol de Gooch. Sin embargo, el estudiante puede filtrar la materia carbonosa por medio de un crisol Gooch ayudado por una succión artificial, lavando la materia carbonosa con un chorro de agua después de que el líquido haya pasado por el filtro. Lavar cuidadosamente el carbón del filtro con agua caliente. Secar el crisol y
el contenido en la estufa de aire a 100 C. La materia carbonosa está ahora lista para la ignición.
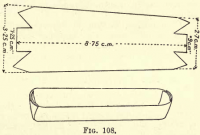
Oxidación del carbono.-Preparar un bote de platino cortando un trozo de lámina de platino como en la fig. 108, doblando los lados y los extremos para formar una cubeta. Transfiera la materia carbonosa y el amianto del Gooch al bote.
El horno de combustión, los accesorios y los aparatos deben ser puestos en orden. El aparato de purificación de oxígeno se utiliza de nuevo, pero esta vez está provisto de un tubo de tres vías, con grifos insertados entre el almacenamiento y los purificadores. Esto permite que una corriente de aire sea arrastrada a través del aparato. El tubo de combustión puede ser de vidrio duro de Jena, de porcelana o de platino. Se utilizan dos tubos en U entre el horno y los bulbos de potasa. El miembro del primer tubo más cercano al horno contiene CuSO4 anhidro y el otro miembro CuCl anhidro. El segundo tubo en U contiene CaCl2 seco. Estos dos tubos forman el «tren de purificación». El CuCl absorbe cualquier Cl, y las otras sustancias cualquier H2O. Este conjunto servirá para muchas determinaciones. Le siguen los bulbos de potasa y los tubos de protección, y se debe tener a mano un aspirador para extraer una corriente de aire a través del aparato cuando sea necesario. Los bulbos de potasa se cargan con 8E. KHO, y el tubo de protección con CaCl2. Se prueba el horno y los bulbos como se ha descrito anteriormente (véase Carbón y Coque), cargándose el tubo como en el croquis, manteniéndose por el momento el bote en el horno de aire a 100° C.
Cuando todo está listo, habiéndose apagado los quemadores durante algún tiempo, se introduce el bote y el contenido. Se encienden los quemadores desde el extremo delantero, se trabaja gradualmente hacia atrás, y se enciende una corriente lenta de oxígeno de unas dos burbujas por segundo hasta que el tubo esté lleno de oxígeno. Se regula la temperatura hasta que el bote tenga un color rojo apagado, y si la solución en los bulbos muestra signos de retroceso hacia el horno, se aumenta la corriente de oxígeno a tres o cuatro burbujas por segundo.
Desde el momento de poner el bote bastarán unos cincuenta minutos para una combustión completa. Apagar el oxígeno y pasar una corriente de aire durante diez minutos.
Los bulbos de potasa y el tubo de protección se retiran ahora y se pesan, y el carbono se calcula como de costumbre.
(b) Carbono grafítico -El hierro es, por algunos, disuelto en HCl, por otros en HNO3 cuando el carbono grafítico permanece como residuo. Para el arrabio cualquiera de los dos métodos, con cuidado, da buenos resultados, pero para los aceros que contienen grafito Blair recomienda la disolución en ácido nítrico. (Para este método consulte a Blair.)
Pese 5 gms. de barrenas de arrabio. Disolver en 50 c.cs. SE. HCl con ayuda de calor suave. Hervir durante unos minutos. Diluir hasta 100 cs. (casi). Filtrar a través de un crisol Gooch. Lavar bien con agua caliente y luego con E. KHO hirviendo. (Esto disuelve cualquier SiO2.) Lavar de nuevo con agua caliente para eliminar el KHO. Secar el crisol y el contenido.
Estimar el carbono como antes por combustión, y calcular el porcentaje como de costumbre.
(c) Carbono combinado (por diferencia).-Siendo conocidos el carbono total y el carbono grafítico, el carbono combinado se obtiene restando el grafítico del carbono total.
Para los métodos directos de estimación consultar las autoridades mencionadas.
ESTIMACIÓN DEL AZUFRE EN EL HIERRO &ACERO
Existen considerables diferencias de opinión en cuanto al mejor método de estimación del azufre en el hierro y el acero. Se admite que el antiguo método de disolución en agua regia y precipitación con BaCl2 es muy inexacto; pero la disolución lenta en HNO3, con muy poca o ninguna presencia de HCl, seguida de una cuidadosa precipitación con BaCl2 en presencia de un exceso definido de HCl y con el debido cuidado en cuanto al tiempo y las condiciones de precipitación, y las precauciones contra la contaminación del precipitado por el hierro, con esto y con cuidado se obtienen buenos resultados. Blair, por otra parte, recomienda la solución en HCl, el S se desprende como H2S, que se absorbe en una solución (alcalina) de Pb(NO3)2 formando PbS, que se disuelve en HCl + KClO3, y el S se precipita como BaSO4. Para otros métodos, véase Blair, Stillman, Auchy, Crobaugh y Drown. Otro método de uso común es el de la evolución del S como H2S, seguido de la absorción en una solución de cloruro de cadmio. El sulfuro de cadmio precipitado se disuelve en HCl y el S se estima por valoración con una solución de yodo, o más común aún, el H2S se absorbe en agua de Br. y luego se precipita como BaSO4 o se absorbe en NaOH y se valora con yodo; este último es el método favorito. (Ver Blair.) Aquí se da el siguiente método:-
Oxidación por HNO3 (el llamado método de Aqua Regia).-Pesar 5 gms. de barrenas y transferirlas a un vaso profundo de 200 c.c.. Añadir con cuidado unos 40 c.cs. 16E. HNO3, en lotes de unos 10 cs. cada vez, cubriendo el vaso de precipitados con un vidrio de reloj grande y teniendo cuidado de que la acción no sea demasiado violenta. Cuando la acción cese aparentemente, observar si todas las partículas se han disuelto (excepto el carbono). Si no es así, calentar en el baño de arena y añadir 3 ó 4 gotas de HCl 16E. HCl, y calentar hasta que se disuelva.
Cuando la solución esté completa, añadir un poco de Na2CO3 para convertir cualquier H2SO4 en Na2SO4, que no es volátil en la evaporación.
Retirar del baño de arena, y añadir 5 c.cs. de HCl fuerte en exceso de lo necesario para disolver cualquier compuesto de hierro precipitado por el Na2CO3. Filtrar el SiO2, y C. lavar bien. Evaporar hasta la sequedad para que el SiO2 sea insoluble. Incorporar con HCl y evaporar hasta que el Fe2Cl6 empiece a cristalizar. Añadir entonces 5 c.cs. HCl. y filtrar si hay algún residuo. (Si no hay ninguno, no había SiO2 en la solución, y se podría haber omitido la evaporación). Filtrar y lavar cuidadosamente el precipitado en el Gooch, llevando el líquido y los lavados hasta unos 100 c.cs.
Calentar hasta la ebullición. Añadir 10 c.cs. de solución saturada de BaCl2. Hervir durante 30 minutos. Dejar reposar durante la noche. Filtrar a través de un Gooch. Lavar con un poco de E. HCl. y luego con agua. Secar, encender y pesar como de costumbre el BaSO4, que debe ser blanco y no estar contaminado con sales de hierro.
Calcular el porcentaje de S de la forma habitual. Como algunos de los reactivos utilizados pueden contener azufre, debe hacerse un ensayo en blanco, utilizando las mismas cantidades que en el análisis real, y cualquier azufre encontrado se deducirá del resultado anterior.
ESTIMACIÓN DEL SILICIO
El método que se da aquí es el de Drown, y es rápido y exacto. El hierro se disuelve en HNO3, seguido de H2SO4, con evaporación hasta sequedad. Esto es seguido por la solución, dejando el silicio en el residuo como SiO2.
Detalles.-Pesar 2 gms de barrenas, y transferir a un plato de platino o porcelana. Añadir 30 c.cs. 8E. HNO3 Cuando la acción aparentemente cesa, añadir 20 c.cs. 18E. H2SO4, y evaporar. (Blair recomienda una suave ráfaga de aire caliente tocando la superficie del líquido. El aire se calienta haciéndolo pasar por una pequeña espiral de tubo de cobre calentado sobre un bunsen. De este modo se acelera la evaporación y se evita la formación de espirales). Continuar la evaporación hasta que salgan copiosos vapores de SO3. Enfriar y diluir cautelosamente con agua destilada hasta 130 c.c. Calentar hasta que se disuelva todo el FeSO4. Filtrar y lavar primero con un poco de E. HCl y luego con agua caliente. Esta filtración se realiza mejor con un papel de filtro sin cenizas de 7 cm. (comprobar la ceniza encendiendo dos o tres de los papeles). Secar; transferir a un crisol de platino; encender como de costumbre y pesar. Añadir al crisol 5 cs. de H2SO4 fuerte y 5 cs. de HF fuerte. Evaporar cuidadosamente hasta la sequedad, utilizando una ráfaga de aire caliente para acelerar la evaporación. Encender y volver a pesar. Si el H2SO4 y el HF son puros, la diferencia de peso representa el SiO2. Comprobar el H2SO4 y el HF (especialmente este último) evaporando un blanco. Cualquier residuo encontrado debe ser tenido en cuenta.
ESTIMACIÓN DEL FÓSFORO
Aquí, de nuevo, se dan numerosos métodos por parte de diferentes autoridades, la mayoría de ellos dando resultados precisos cuando se siguen cuidadosamente. Los dos métodos más adecuados para el análisis técnico son el método de reducción volumétrica preparado por el subcomité (Sres. Barba, Blair, Drown, Dudley y Shimer) del Comité Internacional de Normas del Acero, EE.UU., y el método de reducción modificado dado por los Sres. Dudley y Pease, Jour. Anal. Appl. Chem., vii. 108. El primer método se discute completamente en Blair’s Analysis of Iron; el segundo método se da aquí.
El hierro se disuelve, y el P se precipita como fosfo-molibdato de amonio. Este se disuelve, y por la acción del Zn y el H2SO4 el MoO3 se reduce, y el líquido reducido se valora con K2Mn2O8 (solución estándar), y a partir del número de c.cs. utilizados se puede calcular el contenido de P.
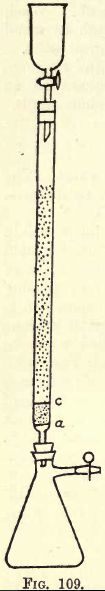
Detalles. -Cuando se va a realizar mucho trabajo, es necesario un aparato de agitación (ver catálogos de suministros químicos). El estudiante, sin embargo, puede realizar la agitación necesaria a mano. Antes de proceder al análisis, debe prepararse el aparato reductor (una modificación del reductor de Jones) (véase la fig. 109).
En a hay un disco finamente perforado de lámina de platino resistente. Entre a y c hay unos ¾ de pulgada de arena blanca limpia, c es otro disco de platino perforado.
Sobre este disco se llena el tubo con zinc amalgamado granulado fino, preparado así:-Disolver 5 gms. Hg en 25 c.cs. de HNO3 fuerte, diluyendo con agua y haciendo la solución hasta 1 litro. En esta solución
verter medio kilo de zinc granulado que pase un tamiz de 20 pero no de 30. Agitar durante uno o dos minutos. Verter la solución. Lavar y secar el zinc, que ahora está amalgamado. El embudo y el matraz se colocan en el aparato como se indica.
Preparar los siguientes reactivos :-
(a) La solución oxidante fuerte de K2Mn2O8. 12 gms. de K2Mn2O8 puro en 1 litro de agua. Filtrar y embotellar.
(b) La Solución de Molibdato.-Disolver 50 gms. MoO3 en 200 c.cs, NH4HO (S.G. .96). Filtrar, y al filtrado añadir 500 c.cs. HNO3 (S.G. 1,2). Dejar reposar al menos 24 horas antes de usar.
(c) La Solución de Sulfato de Amnwnio Ácido.-A 500 c.cs. de agua destilada añadir 27,5 c.cs. NH4HO (S.G. 0.96), y luego 24 c.cs. de H2SO4 puro (S.G. 1.84), y diluir hasta 1000 c.cs.
(d) La solución estándar de K2Mn2O8.-Disolver 2 gms. de K2Mn2O8 cristalizado en 1000 c.cs de agua destilada. Estandarizar la solución de la siguiente manera : Pesar 3 lotes de 0,1 a 0,3 gm. cada uno de alambre de hierro completamente limpio, cuyo contenido de hierro se conoce. Pasar a un matraz Erlenmeyer de 100 c.c. y añadir a cada uno 40 c.c. 8E. H2SO4. Cuando se haya disuelto, hervir 5 minutos; diluir hasta 150 c.c., y pasar por el reductor y lavar, llevando el volumen hasta 200 c.c., como se indica en el análisis. Valorar cada lote con K2Mn2O8. Los resultados deben coincidir con el hierro metálico hasta 1/100 miligramos. Haga la asignación requerida para las impurezas en el alambre tomado. Supongamos que 1 c.c. K2Mn2O8 = .0034923 gm, Fe, entonces multiplique este valor en Fe por la relación de MoO3 a Fe, es decir, .9076, y el producto por la relación del P
presente al MoO3 es decir, .019, tenemos
1 c.c. K2Mn2O8 = .0000602 gm. P
Análisis
Pesar 1 gm. de barrenos. Pasar a un matraz Erlenmeyer de 200 c.c. Añadir 70 cs. 5E. HNO3. Cuando la solución esté completa, hervir un minuto y añadir 10 cs. de la solución «oxidante» de K2Mn2O8. Hervir hasta que desaparezca el color rosa y se separe el MnO2. Retirar y añadir gradualmente con agitación cristales de FeSO4 puro (sin fósforo) hasta que el contenido se aclare. Calentar la solución a 80° C. (si hay As, a 35° C.). Añadir 75 c.c. de la solución de molibdato a una temperatura de 27° C. Cerrar el matraz con un tapón de goma y agitar durante 5 minutos. Dejar reposar durante 5 minutos. Luego filtrar a través de un filtro de 9 cm, y lavar con la solución de sulfato de amonio ácido hasta que unas gotas de los lavados no den color con el sulfuro de amonio.
Disolver el precipitado en el papel con 5 cs. NH4HO (S.G. 0,90) y 25 cs. de agua, dejando que la solución vuelva al matraz original, disolviendo así cualquier precipitado adherido a sus paredes. Lavar hasta que el filtrado y los lavados sumen 150 c.c. Añadir 10 cs. de H2SO4 fuerte (S.G. 1.84) y diluir hasta 200 cs. La solución está ahora lista para la reducción.
Verter 100 c.cs. de H2SO4 caliente ~E/2 en el embudo. Conectar el matraz a la bomba de filtrado y abrir la pinza, de modo que la solución salga casi, pero no del todo, del embudo. A continuación, añadir al embudo el siguiente blanco-5 c.cs. NH4HO (S.G. 0,90), 10 c.cs. H2SO4 (S.G. 1,84), y 50 c.cs. de agua, mezclados entre sí. Abrir de nuevo la abrazadera para que esta mezcla salga casi corriendo del embudo. Añade ahora 200 c.cs, E/2 H2SO4 al embudo, y déjalo correr casi por completo.
Desconecta el matraz, cerrando primero la llave de paso de aceite del embudo. Valorar el contenido del matraz con K2Mn2O8. Generalmente se consumen así unos 0,1 c.c. de permanganato, y esta cantidad debe deducirse de futuras lecturas.
Transfiera ahora la solución a reducir al embudo. Conectar un matraz limpio. Conectar y poner en marcha la bomba del filtro. Abrir la llave de paso y la abrazadera, de forma que casi se pueda pasar la solución. Lavar el matraz que contenía la solución con 100 c.cs. E/2 H2SO4. Añade esto al embudo y trata como antes.
Por último, añade y haz pasar otros 100 cs. del ácido.
La solución reducida en el matraz de filtración debe ser ahora de color verde brillante.
Retira como antes y valora con la solución de permanganato. El verde cambia a marrón rosado, luego a amarillo rosado, después a incoloro, y finalmente se obtiene un rosa permanente (después de permanecer un minuto). A partir de la lectura obtenida dedúzcase la lectura en blanco, y calcúlese el porcentaje de P presente a partir de los datos dados anteriormente.
En lugar de este método volumétrico, algunos químicos prefieren pesar directamente el precipitado amarillo de fosfo-molibdato. Para más detalles, véase Blair’s Analysis of Iron, p. 108.
Nota.-El estudiante debería, siempre que sea posible, aprovechar las referencias de las autoridades especiales. A estas alturas debería ser capaz de consultar, comparar y, hasta cierto punto, utilizar juiciosamente dichos materiales. Ningún libro de texto puede ofrecer un tratamiento exhaustivo del hierro y los aceros, ni de ninguno de los temas tratados en esta sección; por lo tanto, el analista que desee sobresalir en el trabajo técnico debe examinar cuidadosamente las referencias que se ofrecen, junto con la bibliografía actual. Se ha dado la determinación colorimétrica del carbono combinado por el método de Eggertz; el manganeso puede determinarse de manera similar por el método colorimétrico de Peter, o por el método del acetato (véase Blair, etc.).