Dette kapitel indeholder en række oplysninger, som vil give den studerende et indblik i de analysemetoder, der anvendes ved teknisk undersøgelse af jern og stål. Arbejdets karakter forhindrer en detaljeret behandling af emnet – en behandling, der kan kræve en vurdering af et eller flere af følgende stoffer: – Kulstof (frit og kombineret), svovl, silicium, fosfor, mangan, titan, kobber, nikkel, kobolt, chrom, aluminium, arsen, antimon, tin, wolfram, vanadium, kvælstof, jern. Generelt er de mest krævede beregninger af kulstof, svovl, silicium og fosfor de mest nødvendige. Af de andre nævnte grundstoffer og forbindelser er det nødvendigt at bestemme et eller flere af dem for specialstål. For oplysninger om disse bestemmelser henvises den studerende til Chemical Analysis and Foundry Chemistry, af Crobaugh; The. Chemical Analysis of Iron, af Blair ; “Carbon in Steel by Direct Combustion”, af Blount, i The Analyst, Jan. 1902; “Sulphur in Wrought Iron and Steel”, af Auchy, i Jour. Amer. Chem. Soc., marts 1901, og andre artikler i de samme tidsskrifter. Den studerende, der ønsker at gå videre, bør, hvis det er muligt, få adgang til Campbell, Drowns og andres afhandlinger og artikler, som fra tid til anden offentliggøres i de forskellige kemiske og metallurgiske tidsskrifter.
Da den studerendes tid er begrænset, kan han indtil videre udsætte vurderingen af silicium og fosfor, selv om disse er angivet på grund af deres betydning både for metallurg og støber.
For at den studerende kan få en mere grundig forståelse af emnet, vil nogle få bemærkninger om sammensætningen og egenskaberne af de behandlede stoffer ikke være uhensigtsmæssige. Med hensyn til de forskellige grundstoffers indflydelse på stål henvises til The Manufacture and Properties of Structural Steel, af H. H. Campbell.
Kulstof findes i jern i tre tilstande – grafitisk, opløst og kombineret. Udover disse er andre former blevet identificeret ved hjælp af mikroskopet.
Svovl findes i jern hovedsageligt som sulfidet FeS. som er opløseligt i smeltet jern.
Fosfor findes som fosforid af jern, som er fuldstændig opløseligt i det smeltede jern.
Silicium danner silicid af jern, som også er opløseligt i smeltet jern.
Af disse fire grundstoffer er kulstof altså det eneste, der kan eksistere i fri tilstand. Variationerne i proportionerne af de forskellige tilstedeværende grundstoffer er næsten uendelige, men følgende kortfattede tabel giver den omtrentlige sammensætning af råjern, smedejern og stål, skønt hver af disse er genstand for betydelige variationer.
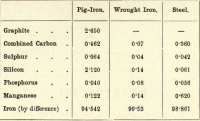
Den studerende skal anslå følgende:-
(1) Kulstof,
(a) Samlet.
(b) Grafitisk.
(c) Kombineret.
(2) Svovl.
(3) Silicium. (Hvis tiden tillader det.)
(4) Fosfor. (Hvis tiden tillader det.)
KARBON I alt
I dette skøn omdannes kulstoffet til CO2, som optages i kaustisk kaliumchlorid. Ud fra vægten af den således opnåede CO2 beregnes kulstoffet.
Af første øjekast ser det ud til, at den enkleste fremgangsmåde ville være at antænde jern- eller stålboringerne direkte i en iltstrøm og absorbere den således dannede CO2 i KHO. Desværre har denne metode hidtil enten vist sig at være upræcis, eller i de tilfælde, hvor der blev opnået fuldstændig forbrænding, var det apparat, der var nødvendigt for at modstå den høje temperatur eller andre variationer i behandlingen, ikke egnet til teknisk arbejde (se artikler af Blount i The Analyst). Den studerende vil opdage, at den her angivne metode på ingen måde er ideel ud fra et teknisk synspunkt, hvad angår bekvemmelighed og hurtighed, og der synes at være en sandsynlighed for, at den i den nærmeste fremtid vil blive erstattet af en hurtigere direkte oxidationsmetode.
Metode vedtaget.- Ved henvisning til de mange værker om dette emne vil man finde en stor variation af metoder. Den her beskrevne metode vil med almindelig omhu give nøjagtige resultater. Kort fortalt er den som følger:-
Jernet opløses i en opløsning af dobbelt kalium- og kobberklorid, der er gjort sur med HCl. Det metalliske kobber udfældes og opløses igen; jernet opløses, mens kulstoffet forbliver i suspension. Det opsamles derefter og antændes i forbrændingsovnen med ilt, og den udviklede CO2 vejes.
Løsning af jernet.-Væg 1 g råjernsboringer af. Overføres til et 300 c.c. bægerglas. Der tilsættes 100 c.cs. CuCl2,2KCl,2H2O-opløsning, som fremstilles på følgende måde. 149,1 dele KCl og 170,3 dele krystalliseret CuCl2, 2H2O opløses i vand. Inddampningen og udkrystalliseringen af dobbeltkloridet. 300 g af dobbeltsaltet opløses i destilleret vand. Filtreres gennem antændt asbest og opbevares i glaspropper.
Til bægerglassets indhold tilsættes 7 c.cs. HCl for at gøre opløsningen sur. Der omrøres med mellemrum, indtil opløsningen af jernet er opnået. Bægerglasset og dets indhold stilles mod slutningen af opløsningen på et vandbad ved en temperatur på ca. 60° C. Følgende reaktioner finder sted-Fe + CuCl2 = FeCl2 + Cu og Cu + CuCl2 = 2CuCl. KCl hjælper blot opløsningen af det udfældede kobber. I løbet af ca. 40 minutter efter tilsætningen af de dobbelte klorider skulle opløsningen være næsten fuldstændig og det meste af kobberet opløst. Bægerglassets sider skylles ned med lidt syrnet dobbeltklorid. Til opløsningen tilsættes lidt antændt asbest for at afvikle det kulstofholdige stof og forhindre, at det tilstopper filteret (som anbefalet af Barba).
Til filtrering er specielle platinbåde, der er monteret efter Goochs digelprincip, meget praktiske. Den studerende kan imidlertid filtrere de kulstofholdige stoffer ved hjælp af en Gooch-dykel hjulpet af kunstig sugning, idet de kulstofholdige stoffer skylles ind med en vandstråle, efter at væsken har passeret gennem filteret. Kulstoffet på filteret vaskes omhyggeligt med varmt vand. Tør diglen og
indholdet i luftovn ved 100 C. Kulstoffet er nu klar til antændelse.
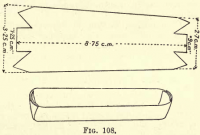
Oxidation af kulstoffet.
Forbered en platinbåd ved at skære et stykke platinfolie ud som i fig. 108 og folde siderne og enderne op, så de danner et trug. Overfør de kulstofholdige stoffer og asbesten fra Gooch’en til båden.
Forbrændingsovnen, tilbehør og fittings skal være bragt i orden. Iltrensningsapparatet anvendes igen, men er denne gang forsynet med et trevejsrør, hvor der er indsat haner mellem lageret og rensningsanlægget. Dette gør det muligt at trække en luftstrøm gennem apparatet. Forbrændingsrøret kan være af hårdt Jena-glas, porcelæn eller platin. Der anvendes to U-rør mellem ovnen og kaliumløg. Den del af det første rør, der er nærmest ovnen, indeholder vandfrit CuSO4 og den anden del vandfrit CuCl. Det andet U-rør indeholder tørret CaCl2. Disse to rør udgør “rensetoget”. CuCl absorberer al Cl, og de andre stoffer absorberer al H2O. Dette sæt vil kunne bruges til mange bestemmelser. Kali- og beskyttelsesrørene følger efter, og der bør være en aspirator til rådighed til at trække en luftstrøm gennem apparatet, når det er nødvendigt. Kaliumlærerne er fyldt med 8E. KHO, og beskyttelsesrøret med CaCl2. Ovn og pærer afprøves som tidligere beskrevet (se Kul og koks), idet røret fyldes som på skitsen, og båden holdes foreløbig i luftovnen ved 100° C.
Når alt er færdigt, idet brænderne har været slukket i nogen tid, indsættes båden og indholdet. Brænderne tændes fra den forreste ende, arbejdes gradvis baglæns, og en langsom iltstrøm på ca. 2 bobler i sekundet er forinden blevet tændt, indtil røret er fuldt af ilt. Regulér temperaturen, indtil båden er matrød, og hvis opløsningen i pærerne viser tegn på at løbe tilbage til ovnen, øges iltstrømmen til tre eller fire bobler pr. sekund.
Fra det tidspunkt, hvor båden sættes i, er ca. halvtreds minutter tilstrækkeligt til en fuldstændig forbrænding. Sluk for ilten og lad en luftstrøm passere i ti minutter.
Kalikuglerne og beskyttelsesrøret fjernes nu og vejes, og kulstoffet beregnes som sædvanligt.
(b) Grafitisk kulstof -Jernet opløses af nogle i HCl, af andre i HNO3, når det grafitiske kulstof forbliver som en rest. For råjern giver begge metoder, med forsigtighed, gode resultater, men for stål, der indeholder grafit, anbefaler Blair opløsning i salpetersyre. (Vedrørende denne metode henvises til Blair.)
Væg 5 gram råjernsboringer ud. Opløses i 50 c.cs. SE. HCl ved hjælp af let varme. Koges i et par minutter. Fortyndes til 100 c.cs. (næsten). Filtreres gennem en Gooch-dæksel. Vask godt med varmt vand og derefter med kogende E. KHO. (Dette opløser eventuelt SiO2.) Vask igen med varmt vand for at fjerne KHO. Diglen og indholdet tørres.
Kulstoffet beregnes som før ved forbrænding, og procentdelen beregnes som sædvanligt.
(c) Kombineret kulstof (ved differens).-Det samlede kulstof og det grafitiske kulstof er kendt, og det kombinerede kulstof fås ved at trække det grafitiske fra det samlede kulstof.
For direkte metoder til estimering henvises til de nævnte autoriteter.
ESTIMERING AF SVEVEL I JERN & STÅL
Der hersker betydelige meningsforskelle med hensyn til den bedste metode til estimering af svovl i jern og stål. Den gamle metode med regnevandsopløsning og BaCl2-udfældning indrømmes at være meget upræcis; men langsom opløsning i HNO3, med meget lidt eller ingen HCl til stede, efterfulgt af omhyggelig udfældning med BaCl2 i nærværelse af et bestemt overskud af HCl og med behørig omhu med hensyn til tid og betingelser for udfældning og forholdsregler mod forurening af udfældningen med jern – med disse og omhu kan man opnå gode resultater. Blair anbefaler derimod opløsning i HCl, idet S udvikles som H2S, der absorberes i en (alkalisk) opløsning af Pb(NO3)2 og danner PbS, som opløses i HCl + KClO3, og S udfældes som BaSO4. For yderligere metoder se Blair, Stillman, Auchy, Crobaugh og Drown. En anden almindelig anvendt metode er udvikling af S i form af H2S efterfulgt af absorption i cadmiumchloridopløsning. Det udfældede cadmiumsulfid opløses i HCl, og S-indholdet bestemmes ved titrering med en jodopløsning, eller, endnu mere almindeligt, H2S absorberes i brintvand og udfældes derefter som BaSO4 eller absorberes i NaOH og titreres med jod; sidstnævnte er den foretrukne metode. (Se Blair.) Følgende metode er her angivet:-
Oxidation med HNO3 (den såkaldte Aqua Regia-metode): – 5 g boringer afvejes og overføres til et dybt 200 c.c.-bægerglas. Tilsæt forsigtigt ca. 40 c.cs. 16E. HNO3 i portioner på ca. 10 c.c. ad gangen, idet bægerglasset dækkes med et stort urglas og man sørger for, at virkningen ikke bliver for voldsom. Når virkningen tilsyneladende ophører, noteres det, om alle partikler er opløst (bortset fra kulstof). Hvis det ikke er tilfældet, opvarmes det på sandbadet, og der tilsættes 3 eller 4 dråber 16E. HCl, og varm op, indtil det er opløst.
Når opløsningen er fuldstændig, tilsættes lidt Na2CO3 for at omdanne eventuel H2SO4 til Na2SO4, som ikke er flygtigt ved inddampning.
Optag sandbadet, og tilsæt 5 c.c. stærk HCl ud over det, der er nødvendigt for netop at opløse eventuelle jernforbindelser, der er udfældet af Na2CO3. SiO2 filtreres fra, og C. Vaskes godt. Inddampes til tørhed for at gøre SiO2 uopløseligt. Der optages HCl, og der inddampes, indtil Fe2Cl6 begynder at udkrystallisere. Derefter tilsættes 5 c.cs. HCl. og filtrer, hvis der er en rest tilbage. (Hvis der ikke er nogen, var der ikke SiO2 i opløsningen, og inddampningen kunne have været udeladt). Udfældningen filtreres og vaskes omhyggeligt i Gooch, idet væske og vaskevand bringes op til ca. 100 c.cs.
Varme op til kogepunktet. Der tilsættes 10 c.cs. mættet opløsning af BaCl2. Koges i 30 minutter. Lad det stå natten over. Filtreres gennem en Gooch. Vask med lidt E. HCl. og derefter med vand. Tør, antændes og vejes som sædvanligt BaSO4, som skal være hvidt og ikke forurenet med jernsalte.
Beregn procentdelen af S på sædvanlig måde. Da nogle af de anvendte reagenser kan indeholde svovl, skal der foretages en blindprøve med de samme mængder som ved den egentlige analyse, og eventuelt fundet svovl trækkes fra det tidligere resultat.
ESTIMERING AF SILIKON
Den her angivne metode er Drowns og er både hurtig og nøjagtig. Jernet opløses i HNO3, efterfulgt af H2SO4 og inddampes til tørhed. Dette efterfølges af opløsning, hvorved siliciumet efterlades i restproduktet som SiO2.
Detaljer: – Der afvejes 2 g boringer og overføres til et platin- eller porcelænsskål. Der tilsættes 30 c.cs. 8E. HNO3 Når virkningen tilsyneladende ophører, tilsættes 20 c.cs. 18E. H2SO4 og inddampes. (Blair anbefaler, at man lader en let varm luftstød spille på væskens overflade. Luften opvarmes ved at lade den passere gennem en lille spiral af kobberrør, der opvarmes over en bunsen. Herved fremskyndes fordampningen, og spirting forhindres). Fortsæt inddampningen, indtil der kommer rigelige dampe af SO3 ud. Afkøles, og fortyndes forsigtigt med destilleret vand til 130 c.cs. Varm op, indtil alt FeSO4 er opløst. Filtrer, og vask først med lidt E. HCl og derefter med varmt vand. Denne filtrering udføres bedst med et 7 cm. askeløst filterpapir (kontrollér asken ved at antænde to eller tre af papirerne). Tørres; overføres til en platindegel; antændes som sædvanligt og vejes. Til smeltediglen tilsættes 5 c.c. stærkt H2SO4 og 5 c.c. stærkt HF. Inddamp forsigtigt til tørhed ved hjælp af en varm luftstorm for at fremskynde inddampningen. Der antændes, og der vejes igen. Forudsat at H2SO4 og HF er rene, repræsenterer vægtforskellen SiO2. H2SO4 og HF (især sidstnævnte) kontrolleres ved inddampning af en blindprøve. Der skal tages hensyn til en eventuel rest.
ESTIMATION AF PHOSFOR
Også her er der angivet mange metoder af forskellige autoriteter, og de fleste af dem giver nøjagtige resultater, når de følges nøje. De to metoder, der er mest velegnede til teknisk analyse, er den volumetriske reduktionsmetode, der er udarbejdet af underudvalget (de herrer Barba, Blair, Drown, Dudley og Shimer) under International Steel Standards Committee, U.S.A., og den modificerede reduktionsmetode, som er givet af de herrer Dudley og Pease, Jour. Anal. Appl. Chem., vii. 108. Førstnævnte metode er udførligt beskrevet i Blair’s Analysis of Iron; sidstnævnte metode er angivet her.
Jernet opløses, og P’et udfældes som phospho-molybdat af ammonium. Dette opløses, og ved virkningen af Zn og H2SO4 reduceres MoO3, og den reducerede væske titreres derefter med K2Mn2O8 (standardopløsning), og ud fra det anvendte antal c.c.c. kan P-indholdet beregnes.
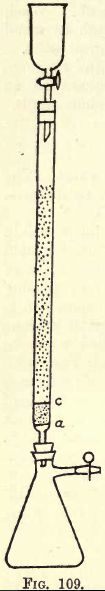
Detaljer. -Hvis der skal arbejdes meget, er det nødvendigt med et omrystningsapparat (se kemikalieforsyningskataloger). Den studerende kan dog selv foretage den nødvendige omrystning i hånden. Før analysen foretages, skal reduktionsapparatet (en modifikation af Jones-reduktionsapparatet) forberedes (se fig. 109).
På a er der en fint perforeret skive af kraftig platinfolie. Mellem a og c er der ca. ¾ tomme rent hvidt sand, c er en anden perforeret platinskive.
Over denne skive fyldes røret med fint granuleret amalgameret zink, der er fremstillet således: – Opløs 5 g. Hg i 25 c.cs. stærkt HNO3, der fortyndes med vand og fyldes op til 1 liter. I denne opløsning
hældes et halvt kilo granuleret zink, der passerer en 20, men ikke en 30 sigte. Rystes i et eller to minutter. Opløsningen hældes fra. Zinken, som nu er amalgameret, vaskes og tørres. Tragt og kolbe monteres på apparatet som vist.
Forbered følgende reagenser :-
(a) Den stærkt oxiderende opløsning af K2Mn2O8. 12 g ren K2Mn2O8 i 1 liter vand. Filtrer og aftap på flaske.
(b) Molybdatopløsning: Opløs 50 g. MoO3 i 200 c.cs, NH4HO (S.G. .96). Filtrer, og til filtratet tilsættes 500 c.cs. HNO3 (S.G. 1,2). Lad det stå mindst 24 timer før brug.
(c) Den sure amnwniumsulfatopløsning.- Til 500 c.c. destilleret vand tilsættes 27,5 c.c. NH4HO (S.G. 0,96) og derefter 24 c.cs. rent H2SO4 (S.G. 1,84) og fortyndes til 1000 c.cs.
(d) Standard K2Mn2O8-opløsning.- 2 g krystalliseret K2Mn2O8 opløses i 1000 c.cs. destilleret vand. Opløsningen standardiseres som følger : Der afvejes 3 partier på hver 0,1 til 0,3 g grundigt renset jerntråd, hvis jernindhold er kendt. De overføres til 100 c.c. Erlenmeyer-kolber og tilsættes hver 40 c.cs. 8E. H2SO4. Når de er opløst, koges de i 5 minutter; de fortyndes til 150 ccm og passeres gennem reduktionsapparatet og vaskes, hvorved mængden bringes op til 200 ccm, som angivet i analysen. Hvert parti titreres med K2Mn2O8. Resultaterne skal stemme overens med hensyn til metallisk jern med en nøjagtighed på 1/100 milligram. Der skal tages hensyn til urenhederne i den udtagne tråd. Hvis man antager, at 1 c.c. K2Mn2O8 = 0,0034923 gm Fe, multiplicerer man denne værdi i Fe med forholdet mellem MoO3 og Fe, nemlig 0,9076, og produktet med forholdet mellem P
til stede og MoO3, nemlig 0,019, så får man
1 c.c. K2Mn2O8 = 0,0000602 gm. P
Analyse
Væg 1 gm. boringer. Overføres til en 200 c.c. Erlenmeyer-kolbe. Tilsæt 70 c.cs. 5E. HNO3. Når opløsningen er færdig, koges den et minut, og der tilsættes 10 c.c. af den “oxiderende” opløsning af K2Mn2O8. Koges, indtil den lyserøde farve forsvinder, og MnO2 udskilles. Den fjernes, og der tilsættes gradvis under omrøring krystaller af rent (fosforfrit) FeSO4, indtil indholdet er klart. Opløsningen opvarmes til 80° C. (hvis der er As til stede, til 35° C.). Der tilsættes 75 c.c. molybdatopløsning ved en temperatur på 27° C. Kolben lukkes med en gummiprop og rystes i 5 minutter. Lad den stå i 5 minutter. Derefter filtreres gennem et 9 cm, filtreres og vaskes med den sure amm. sulfatopløsning, indtil nogle få dråber af vaskevandet ikke giver nogen farve med ammoniumsulfid.
Udløs udfældningen på papiret med 5 c.cs. NH4HO (S.G. 0,90) og 25 c.c. vand, idet man lader opløsningen løbe tilbage i den oprindelige kolbe, hvorved udfældningen, der måtte klæbe til siderne, opløses. Der vaskes, indtil filtrat og vaskevand udgør 150 c.cs. Der tilsættes 10 c.cs. stærkt H2SO4 (S.G. 1,84), og der fortyndes til 200 c.cs. Opløsningen er nu klar til reduktion.
Hæld 100 c.cs. varmt ~E/2 H2SO4 i tragten. Kolben tilsluttes filterpumpen, og klemmen åbnes, således at opløsningen næsten, men ikke helt, løber ud af tragten. I tragten tilsættes derefter følgende blanco-5 c.cs. NH4HO (S.G. 0,90), 10 c.cs. H2SO4 (S.G. 1,84) og 50 c.c. vand, der blandes sammen. Klemmen åbnes igen, således at denne blanding næsten løber ud af tragten. Der tilsættes nu 200 c.cs, E/2 H2SO4 til tragten, og den løber næsten igennem.
Kolben afbrydes, idet man først lukker proppen olie tragten. Kolbens indhold titreres med K2Mn2O8. I almindelighed forbruges der herved ca. 0,1 c.cs. permanganat, og denne mængde skal fratrækkes ved fremtidige aflæsninger.
Nu overføres den opløsning, der skal reduceres, til tragten. Der anbringes en ren kolbe. Filterpumpen tilsluttes og sættes i gang. Åbn hanen og klemmen, således at opløsningen næsten løber igennem. Kolben, der indeholdt opløsningen, skylles ud med 100 c.cs. E/2 H2SO4. Dette tilsættes til tragten og behandles som før.
Sidst tilsættes og næsten gennemløbes endnu 100 c.cs. af syren.
Den reducerede opløsning i filterkolben skal nu være lysegrøn.
Fjernes som før og titreres med permanganatopløsningen. Den grønne farve ændrer sig til lyserødbrun, derefter lysegul, derefter farveløs, og til sidst fås en permanent lyserød farve (efter 1 minuts standtid). Ud fra den opnåede aflæsning trækkes blancoaflæsningen fra, og procentdelen af tilstedeværende P beregnes ud fra de ovenfor anførte data.
I stedet for denne volumetriske metode foretrækker nogle kemikere at veje det gule fosfomolybdatudfældning direkte. For nærmere oplysninger se Blair’s Analysis of Iron, s. 108.
Note: – Den studerende bør, hvor det er muligt, benytte sig af henvisninger til særlige autoriteter. På dette tidspunkt bør han være i stand til at konsultere, sammenligne og i en vis udstrækning at anvende sådanne materialer med omtanke. Ingen enkelt lærebog kan på nogen måde give en omfattende behandling af “Jern og stål” eller for den sags skyld af noget af de emner, der behandles i dette afsnit; derfor skal sådanne henvisninger, som er givet, sammen med den aktuelle litteratur, omhyggeligt gennemgås af den analytiker, der ønsker at udmærke sig i teknisk arbejde. Den kolorimetriske bestemmelse af kombineret kulstof ved Eggertz’ metode er givet ; mangan kan bestemmes noget tilsvarende ved Peters kolorimetriske metode eller ved acetatmetoden (se Blair, etc.).