On présente dans ce chapitre ce qui donnera à l’étudiant un aperçu des méthodes d’analyse utilisées dans l’examen technique du fer et de l’acier. La nature de cet ouvrage empêche un traitement détaillé du sujet, traitement qui peut exiger une estimation de l’une ou de plusieurs des substances suivantes : carbone (libre et combiné), soufre, silicium, phosphore, manganèse, titane, cuivre, nickel, cobalt, chrome, aluminium, arsenic, antimoine, étain, tungstène, vanadium, azote, fer. En général, les estimations les plus nécessaires sont celles du carbone, du soufre, du silicium et du phosphore. Parmi les autres éléments et composés mentionnés, la détermination d’un ou plusieurs d’entre eux est nécessaire dans le cas des aciers spéciaux. Pour obtenir des renseignements sur ces déterminations, l’étudiant est invité à consulter Chemical Analysis and Foundry Chemistry, de Crobaugh ; The. Chemical Analysis of Iron, par Blair ; « Carbon in Steel by Direct Combustion, » par Blount, dans The Analyst, Jan. 1902 ; « Sulphur in Wrought Iron and Steel, » par Auchy, dans le Jour. Amer. Chem. Soc., mars 1901, et d’autres articles dans les mêmes revues. L’étudiant qui désire aller plus loin devrait, si possible, obtenir l’accès aux papiers et articles de Campbell, Drown, et autres, publiés de temps à autre dans les divers journaux chimiques et métallurgiques.
Comme le temps de l’étudiant est limité, il peut pour le moment remettre à plus tard l’estimation du silicium et du phosphore, bien que ceux-ci soient donnés en raison de leur importance à la fois pour le métallurgiste et le fondeur.
Afin que l’étudiant puisse obtenir une compréhension plus approfondie du sujet, quelques notes 0sur la composition et les propriétés des substances considérées ne seront pas hors de propos. En ce qui concerne l’influence des divers éléments sur l’acier, consulter The Manufacture and Properties of Structural Steel, de H. H. Campbell.
Le carbone existe dans le fer sous trois états : graphitique, dissous et combiné. En plus de ceux-ci, d’autres formes ont été identifiées par le microscope.
Le soufre existe dans le fer principalement sous forme de sulfure FeS. qui est soluble dans le fer fondu.
Le phosphore existe sous forme de phosphure de fer, qui est complètement soluble dans le fer fondu.
Le silicium forme du siliciure de fer, qui est également soluble dans le fer fondu.
De ces quatre éléments, le carbone est donc le seul qui puisse exister à l’état libre. Les variations dans les proportions des différents éléments présents sont presque infinies, mais le bref tableau suivant donne la composition approximative de la fonte brute, du fer forgé et de l’acier, bien que chacun de ces éléments soit sujet à des variations considérables.
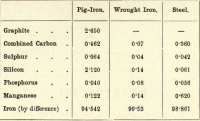
L’étudiant doit estimer les éléments suivants:-
(1) Carbone,
(a) Total.
(b) Graphitique.
(c) Combiné.
(2) Soufre.
(3) Silicium. (Si le temps le permet.)
(4) Phosphore. (Si le temps le permet.)
CARBONE Total
Dans cette estimation, le carbone est transformé en CO2 qui est absorbé par la potasse caustique. A partir du poids de CO2 ainsi obtenu, on calcule le carbone.
A première vue, il semblerait que le procédé le plus simple serait d’enflammer les forages en fer ou en acier directement dans un courant d’oxygène et d’absorber le CO2 ainsi formé dans la KHO. Malheureusement, cette méthode, jusqu’à présent, s’est avérée inexacte ou, lorsque la combustion complète a été obtenue, l’appareil nécessaire pour résister à la température élevée ou à d’autres variations de traitement n’était pas adapté au travail technique (voir les articles de Blount dans The Analyst). L’étudiant constatera que la méthode donnée ici est loin d’être idéale, du point de vue technique, sur le plan de la commodité et de la rapidité, et il semble probable qu’elle sera remplacée dans un proche avenir par quelque méthode d’oxydation directe plus rapide.
Méthode adoptée.- En se référant aux nombreux ouvrages sur ce sujet, on trouvera une grande variété de méthodes. La méthode donnée ici donnera, avec un soin ordinaire, des résultats précis. Brièvement, elle est la suivante:-
Le fer est dissous dans une solution de chlorure double de potassium et de cuivre, rendue acide par HCl. Le cuivre métallique est précipité et redissous ; le fer est dissous, le carbone étant laissé en suspension. On le recueille alors et on l’enflamme dans le fourneau de combustion avec de l’oxygène, et on pèse le CO2 dégagé.
Solution du fer.-Peser 1 gramme de forures de fonte brute. Transférer dans un bécher de 300 c.c.. Ajouter 100 c.cs. solution de CuCl2,2KCl,2H2O, qui se prépare comme suit. Dissoudre dans l’eau 149,1 parties de KCl et 170,3 parties de CuCl2, 2H2O cristallisé. Evaporer et cristalliser le chlorure double. Dissoudre 300 g du sel double dans de l’eau distillée. Filtrer à travers de l’amiante enflammé, et conserver dans des bouteilles en verre bouchées.
Au contenu du bécher, ajouter 7 c.cs. HCl pour rendre la solution acide. Remuer par intermittence jusqu’à ce que la solution du fer soit effectuée. Placez le bécher et son contenu vers la fin de la solution sur un bain-marie à une température d’environ 60° C. Les réactions suivantes ont lieu : Fe + CuCl2 = FeCl2 + Cu et Cu + CuCl2 = 2CuCl. Le KCl facilite simplement la mise en solution du cuivre précipité. Dans environ 40 minutes après l’ajout des chlorures doubles, la solution devrait être presque complète et la plupart du cuivre dissous. Lavez les parois du bécher avec un peu de double chlorure acidulé. Ajouter à la solution un peu d’amiante enflammé pour décanter les matières carbonées et éviter qu’elles n’obstruent le filtre (selon les recommandations de Barba).
Pour la filtration, des bateaux spéciaux en platine, montés sur le principe du creuset de Gooch, sont très commodes. L’étudiant peut cependant filtrer la matière carbonée au moyen d’un creuset de Gooch aidé d’une aspiration artificielle, la matière carbonée étant lavée par un jet d’eau après le passage du liquide dans le filtre. Laver soigneusement à l’eau chaude le carbone présent sur le filtre. Séchez le creuset et
les contenus dans l’étuve à air à 100 C. La matière carbonée est maintenant prête à être enflammée.
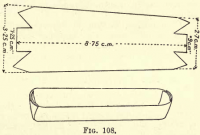
Oxidation du carbone.-Préparez une nacelle de platine en découpant un morceau de feuille de platine comme dans la figure 108, en repliant les côtés et les extrémités pour former une auge. Transférer les matières carbonées et l’amiante du Gooch dans la nacelle.
Le fourneau de combustion, les accessoires et la robinetterie doivent être mis en ordre. On utilise à nouveau l’appareil d’épuration de l’oxygène, mais cette fois, il est muni d’un tube à trois voies, avec des robinets insérés entre le stockage et les épurateurs. Cela permet d’aspirer un courant d’air à travers l’appareil. Le tube de combustion peut être en verre dur de Jena, en porcelaine ou en platine. Deux tubes en U sont utilisés entre le four et les bulbes de potasse. La branche du premier tube la plus proche du four contient du CuSO4 anhydre, l’autre branche du CuCl anhydre. Le second tube en U contient du CaCl2 séché. Ces deux tubes forment le « train de purification ». Le CuCl absorbe tout Cl, et les autres substances tout H2O. Cet ensemble servira pour de nombreuses déterminations. Les ampoules de potasse et les tubes de garde suivent, et un aspirateur doit être à portée de main pour aspirer un courant d’air à travers l’appareil lorsque cela est nécessaire. Les ampoules de potasse sont chargées avec 8E. KHO, et le tube de garde avec du CaCl2. Testez le four et les ampoules comme décrit précédemment (voir Charbon et coke), le tube étant chargé comme dans le croquis, la nacelle étant pour le moment maintenue dans le four à air à 100° C.
Quand tout est prêt, les brûleurs ayant été éteints pendant un certain temps, la nacelle et son contenu sont insérés. Les brûleurs sont allumés à partir de l’extrémité avant, travaillent progressivement vers l’arrière, et un courant lent d’oxygène d’environ deux bulles par seconde ayant été précédemment allumé jusqu’à ce que le tube soit plein d’oxygène. Réglez la température jusqu’à ce que le bateau soit à un rouge terne, et si la solution dans les ampoules montre des signes de reflux vers le fourneau, augmentez le courant d’oxygène à trois ou quatre bulles par seconde.
Depuis le moment où l’on met le bateau, environ cinquante minutes suffiront pour une combustion complète. Arrêtez l’oxygène et faites passer un courant d’air pendant dix minutes.
Les ampoules de potasse et le tube de garde sont maintenant retirés et pesés, et le carbone calculé comme d’habitude.
(b) Le carbone graphitique -Le fer est, par certains, dissous dans HCl, par d’autres dans HNO3 lorsque le carbone graphitique reste comme un résidu. Pour la fonte brute, l’une ou l’autre méthode, si l’on fait attention, donne de bons résultats, mais pour les aciers contenant du graphite, Blair recommande la solution dans l’acide nitrique. (Pour cette méthode, consulter Blair.)
Peser 5 g de fer brut. Dissoudre dans 50 c.cs. SE. HCl à l’aide d’une chaleur douce. Faites bouillir pendant quelques minutes. Diluer à 100 c.cs. (presque). Filtrer à travers un creuset de Gooch. Lavez bien avec de l’eau chaude et ensuite avec de l’E. KHO bouillant. (Lavez à nouveau à l’eau chaude pour éliminer le KHO. Séchez le creuset et le contenu.
Estimez le carbone comme précédemment par combustion, et calculez le pourcentage comme d’habitude.
(c) Carbone combiné (par différence).-Le carbone total et le carbone graphitique étant connus, le carbone combiné est obtenu en soustrayant le graphitique du carbone total.
Pour les méthodes directes d’estimation, consulter les autorités mentionnées.
ESTIMATION DU SOUFRE DANS LE FER& ACIER
Des divergences d’opinion considérables existent quant à la meilleure méthode d’estimation du soufre dans le fer et l’acier. On admet que l’ancienne méthode de la solution d’eau régale et de la précipitation du BaCl2 est très inexacte ; mais une solution lente dans HNO3, avec très peu ou pas de HCl présent, suivie d’une précipitation soigneuse par BaCl2 en présence d’un excès défini de HCl et avec le soin nécessaire quant au temps et aux conditions de la précipitation, et les précautions contre la contamination du précipité par le fer – avec ces précautions et ce soin, on obtient de bons résultats. Blair, par contre, recommande une solution dans HCl, le S étant dégagé sous forme de H2S, qui est absorbé dans une solution (alcaline) de Pb(NO3)2 formant PbS, qui est dissous dans HCl + KClO3, et le S précipité sous forme de BaSO4. Pour d’autres méthodes, voir Blair, Stillman, Auchy, Crobaugh et Drown. Une autre méthode couramment utilisée est celle de l’évolution du S sous forme de H2S, suivie d’une absorption dans une solution de chlorure de cadmium. Le sulfure de cadmium précipité est dissous dans HCl et le S est estimé par titrage avec une solution d’iode, ou encore, ce qui est plus courant, le H2S est absorbé dans l’eau de Br. puis précipité sous forme de BaSO4 ou absorbé dans NaOH et titré avec de l’iode ; cette dernière méthode est la plus utilisée. (Voir Blair.) La méthode suivante est donnée ici:-
Oxidation par HNO3 (la méthode dite Aqua Regia).-Peser 5 gms. de borings et les transférer dans un bécher profond de 200 c.c.. Ajouter avec précaution environ 40 c.cs. 16E. HNO3, par lots d’environ 10 c.c. à la fois, en couvrant le bécher d’un grand verre de montre et en veillant à ce que l’action ne soit pas trop violente. Lorsque l’action semble cesser, notez si toutes les particules sont dissoutes (sauf le carbone). Si ce n’est pas le cas, chauffez sur le bain de sable, et ajoutez 3 ou 4 gouttes de 16E. HCl, et chauffer jusqu’à dissolution.
Lorsque la solution est complète, ajouter un peu de Na2CO3 pour convertir tout H2SO4 en Na2SO4, qui n’est pas volatil à l’évaporation.
Retirer du bain de sable, et ajouter 5 c.c. de HCl fort en excès de ce qui est nécessaire pour juste dissoudre tout composé de fer précipité par le Na2CO3. Filtrez le SiO2, et C. Lavez bien. Évaporer à sec pour rendre le SiO2 insoluble. Reprendre avec HCl et évaporer jusqu’à ce que le Fe2Cl6 commence à cristalliser. Ajouter alors 5 c.cs. HCl. et filtrer si un résidu est présent. (S’il n’y en a pas, c’est qu’il n’y avait pas de SiO2 en solution, et l’évaporation aurait pu être omise). Filtrer et laver soigneusement le précipité dans le Gooch, en portant le liquide et les lavages à environ 100 c.cs.
Chauffer jusqu’à ébullition. Ajouter 10 c.cs. de solution saturée de BaCl2. Faire bouillir pendant 30 minutes. Laisser reposer pendant la nuit. Filtrer à travers un Gooch. Laver avec un peu de E. HCl. et ensuite avec de l’eau. Sécher, enflammer et peser comme d’habitude le BaSO4, qui doit être blanc, et non contaminé par des sels de fer.
Calculer le pourcentage de S de la manière habituelle. Comme certains des réactifs utilisés peuvent contenir du soufre, il faut faire un essai à blanc, en utilisant les mêmes quantités que pour l’analyse réelle, et tout soufre trouvé doit être déduit du résultat précédent.
ESTIMATION DU SILICIUM
La méthode donnée ici est celle de Drown, et est à la fois rapide et exacte. Le fer est dissous dans HNO3, suivi de H2SO4, avec évaporation à sec. On procède ensuite à la mise en solution, en laissant le silicium dans le résidu sous forme de SiO2.
Détails.-Peser 2 gms de borings, et les transférer dans un plat de platine ou de porcelaine. Ajouter 30 c.cs. 8E. HNO3 Lorsque l’action semble cesser, ajouter 20 c.cs. 18E. H2SO4, et évaporer. (Blair recommande un léger souffle d’air chaud jouant sur la surface du liquide. L’air est chauffé en le faisant passer à travers une petite spirale de tuyau de cuivre chauffé sur un bunsen. L’évaporation est ainsi accélérée et la spirale évitée). Continuez l’évaporation jusqu’à ce que de copieuses fumées de SO3 se dégagent. Refroidissez, et diluez prudemment avec de l’eau distillée à 130 c.cs. Chauffez jusqu’à ce que tout le FeSO4 soit dissous. Filtrez, et lavez d’abord avec un peu de E. HCl, et ensuite avec de l’eau chaude. Cette filtration s’effectue de préférence avec un papier filtre de 7 cm. sans cendres (vérifiez les cendres en enflammant deux ou trois des papiers). Sécher ; transférer dans un creuset en platine ; enflammer comme d’habitude et peser. Dans le creuset, ajoutez 5 c.c. de H2SO4 fort et 5 c.c. de HF fort. Évaporez soigneusement jusqu’à siccité, en utilisant un souffle d’air chaud pour accélérer l’évaporation. Enflammer et peser à nouveau. A condition que le H2SO4 et le HF soient purs, la différence de poids représente du SiO2. Vérifiez le H2SO4 et le HF (en particulier ce dernier) en évaporant un blanc. Tout résidu trouvé doit être pris en compte.
ESTIMATION DU PHOSPHORE
Ici encore, de nombreuses méthodes sont données par différentes autorités, la majorité d’entre elles donnant des résultats précis lorsqu’elles sont suivies avec soin. Les deux méthodes les mieux adaptées à l’analyse technique sont la méthode de réduction volumétrique préparée par le sous-comité (MM. Barba, Blair, Drown, Dudley et Shimer) de l’International Steel Standards Committee, U.S.A., et la méthode de réduction modifiée donnée par MM. Anal. Appl. Chem. vii. 108. La première méthode est entièrement discutée dans Blair’s Analysis of Iron ; la seconde méthode est donnée ici.
Le fer est dissous, et le P précipité sous forme de phospho-molybdate d’ammonium. Celui-ci est dissous, et par l’action de Zn et de H2SO4, le MoO3 est réduit, et le liquide réduit est ensuite titré avec K2Mn2O8 (solution standard), et à partir du nombre de c.c. utilisés, on peut calculer la teneur en P.
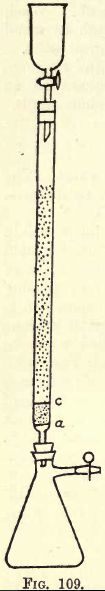
Détails. -Lorsqu’un travail important doit être effectué, un appareil d’agitation est nécessaire (voir les catalogues de fournitures chimiques). L’étudiant, cependant, peut effectuer l’agitation nécessaire à la main. Avant de procéder à l’analyse, l’appareil réducteur (une modification du réducteur de Jones) doit être préparé (voir fig. 109).
Au niveau de a se trouve un disque finement perforé de feuille de platine robuste. Entre a et c, il y a environ ¾ de pouce de sable blanc propre, c est un autre disque de platine perforé.
Au-dessus de ce disque, le tube est rempli de zinc amalgamé à granulés fins, préparé ainsi:-Dissoudre 5 gms. Hg dans 25 c.cs. de HNO3 fort, en diluant avec de l’eau et en portant la solution à 1 litre. Dans cette solution, versez un demi-kilo de zinc granulé qui passe par un tamis de 20 mais pas de 30. Agitez pendant une ou deux minutes. Versez la solution. Laver et sécher le zinc, qui est maintenant amalgamé. L’entonnoir et le ballon sont montés sur l’appareil comme indiqué.
Préparez les réactifs suivants :-
(a) La solution fortement oxydante de K2Mn2O8. 12 g de K2Mn2O8 pur dans 1 litre d’eau. Filtrer et mettre en bouteille.
(b) La solution de molybdate.-Dissoudre 50 gms. MoO3 dans 200 c.cs, NH4HO (S.G. .96). Filtrer, et au filtrat ajouter 500 c.cs. HNO3 (S.G. 1.2). Laisser reposer au moins 24 heures avant de l’utiliser.
(c) La solution de sulfate d’amnwnium acide.-A 500 c.c. d’eau distillée, ajouter 27,5 c.cs. NH4HO (S.G. 0,96), puis 24 c.cs. de H2SO4 pur (S.G. 1,84), et diluer à 1000 c.cs.
(d) La solution standard de K2Mn2O8.-Dissoudre 2 gms. de K2Mn2O8 cristallisé dans 1000 c.cs d’eau distillée. Etalonnez la solution comme suit : Peser 3 lots de 0,1 à 0,3 g chacun de fil de fer soigneusement nettoyé, dont la teneur en fer est connue. Transférer dans une fiole Erlenmeyer de 100 c.c. et ajouter à chaque lot 40 c.c. 8E. H2SO4. Une fois dissous, faire bouillir pendant 5 minutes ; diluer à 150 c.c., passer dans le réducteur et laver, en portant le volume à 200 c.c., comme indiqué dans l’analyse. Titrer chaque lot avec du K2Mn2O8. Les résultats doivent concorder pour le fer métallique au 1/100 de milligramme. Faites l’allocation requise pour les impuretés dans le fil pris. Supposons que 1 c.c. de K2Mn2O8 = .0034923 gm, Fe, puis multiplions cette valeur en Fe par le rapport du MoO3 au Fe, soit .9076, et le produit par le rapport du P
présent au MoO3, soit .019, nous avons
1 c.c. de K2Mn2O8 = .0000602 gm. P
Analyse
Peser des forages de 1 gm. Transférer dans une fiole Erlenmeyer de 200 c.c.. Ajouter 70 c.c. 5E. HNO3. Lorsque la solution est complète, faire bouillir une minute et ajouter 10 c.c. de la solution « oxydante » de K2Mn2O8. Faire bouillir jusqu’à ce que la couleur rose disparaisse et que le MnO2 se sépare. Retirer, et ajouter graduellement en agitant des cristaux de FeSO4 pur (sans phosphore) jusqu’à ce que le contenu devienne clair. Chauffer la solution à 80° C. (si As est présent, à 35° C.). Ajouter 75 c.c. de la solution de molybdate à une température de 27° C. Fermer le flacon avec un bouchon en caoutchouc et agiter pendant 5 minutes. Laisser reposer pendant 5 minutes. Filtrer ensuite à travers un filtre de 9 cm, et laver avec la solution acide de sulfate d’ammonium jusqu’à ce que quelques gouttes des lavages ne donnent aucune couleur avec le sulfure d’ammonium.
Dissoudre le précipité sur le papier avec 5 c.cs. NH4HO (S.G. 0,90) et 25 c.cs. d’eau, en laissant la solution s’écouler de nouveau dans la fiole originale, dissolvant ainsi tout précipité adhérant à ses côtés. Laver jusqu’à ce que le filtrat et les lavages atteignent 150 c.cs. Ajouter 10 c.c. de H2SO4 fort (S.G. 1.84), et diluer à 200 c.cs. La solution est maintenant prête pour la réduction.
Versez 100 c.cs. de ~E/2 H2SO4 chaud dans l’entonnoir. Relier la fiole à la pompe à filtre et ouvrir le clamp, de sorte que la solution s’écoule presque, mais pas tout à fait, de l’entonnoir. Ajouter ensuite à l’entonnoir le blanc suivant : 5 c.c. NH4HO (S.G. 0.90), 10 c.cs. H2SO4 (S.G. 1.84), et 50 c.c. d’eau, mélangés ensemble. Ouvrez à nouveau le clapet, de manière à faire presque couler ce mélange hors de l’entonnoir. Ajoutez maintenant 200 c.cs, E/2 H2SO4 dans l’entonnoir, et faites-le presque couler.
Débranchez la fiole, en fermant d’abord le robinet d’arrêt huile l’entonnoir. Titrer le contenu de la fiole avec du K2Mn2O8. Généralement, environ 0,1 c.c. de permanganate sont ainsi consommés, et cette quantité doit être déduite des lectures futures.
Transférez maintenant la solution à réduire dans l’entonnoir. Fixez une fiole propre. Connectez et démarrez la pompe à filtre. Ouvrez le robinet d’arrêt et la pince, de manière à faire presque couler la solution. Laver la fiole qui contenait la solution avec 100 c.cs. E/2 DE H2SO4. Ajoutez cette solution dans l’entonnoir et traitez-la comme précédemment.
Enfin, ajoutez et faites passer presque entièrement 100 c.c. d’acide.
La solution réduite dans la fiole filtrante doit maintenant être d’un vert vif.
Retirez-la comme précédemment et titrez-la avec la solution de permanganate. Le vert se transforme en brun rosé, puis en jaune rosé, puis en incolore, et enfin on obtient un rose permanent (après avoir reposé une minute). De la lecture obtenue déduire la lecture à blanc, et calculer le pourcentage de P présent à partir des données données données ci-dessus.
Au lieu de cette méthode volumétrique, certains chimistes préfèrent peser directement le précipité jaune de phospho-molybdate. Pour plus de détails, voir Blair’s Analysis of Iron, p. 108.
Note.-L’étudiant devrait, dans la mesure du possible, profiter des références aux autorités spéciales. A ce stade, il devrait être capable de consulter, de comparer et, dans une certaine mesure, d’utiliser judicieusement ces matériaux. Aucun manuel ne peut donner un traitement complet du fer et de l’acier ou, d’ailleurs, de n’importe lequel des sujets traités dans cette section ; par conséquent, les références qui sont données, ainsi que la littérature courante, doivent être soigneusement étudiées par l’analyste qui souhaite exceller dans le travail technique. La détermination colorimétrique du carbone combiné par la méthode d’Eggertz a été donnée ; le manganèse peut être déterminé de façon similaire par la méthode colorimétrique de Peter, ou par la méthode de l’acétate (voir Blair, etc.).