Introducción
El metano (CH4) es un importante gas de efecto invernadero. Su emisión desde entornos naturales como humedales, océanos y sedimentos representa más del 70% del metano atmosférico a nivel mundial (IPCC, 2007). Una evaluación de los datos publicados reveló diferentes tasas de emisión de metano para los humedales, los lagos, los ríos, los estuarios y los océanos (en orden decreciente, Figura Suplementaria S1). Los humedales naturales representan por sí solos el 62% de la producción de CH4 biogénico (Kirschke et al., 2013; Nazaries et al., 2013) y las emisiones de los humedales dominan la variabilidad interanual de las fuentes de metano (Bousquet et al., 2006). En cambio, la vasta superficie de los ecosistemas marinos solo contribuye en un 8% a las fuentes naturales de CH4 (Nazaries et al., 2013). Dentro de la emisión global de metano oceánico, menos del 10% es aportado por los estuarios (Bange et al., 1994).
La metanogénesis, la formación biológica de metano, es realizada por arqueas metanogénicas que producen metano principalmente a partir de H2/CO2, grupos metilo o acetato en condiciones anóxicas (Thauer et al., 2008). Esta reacción es catalizada por la metil-coenzima M reductasa (MCR). El gen mcrA que codifica una subunidad de esta enzima es un marcador genético comúnmente utilizado en los estudios moleculares (Conrad, 2007; Bridgham et al., 2013). La ventaja del marcador del gen mcrA es capturar tanto las firmas filogenéticas como funcionales de los metanógenos, ofreciendo una alta profundidad de secuenciación para esta función en particular (Luton et al., 2002; Borrel et al., 2013; Yang et al., 2014). Se recuperó un gran número de secuencias de mcrA de una variedad de entornos naturales. El conjunto de datos públicos de mcrA permite extraer patrones ecológicos generales e investigar la conformación de gradientes ambientales a escala global y regional. Además, existe una base de datos que resume las propiedades fisiológicas de 152 aislados metanogénicos1 (Jabłoński et al., 2015). Recientemente, el binning del genoma reveló secuencias mcrA inusuales en la nueva clase de Methanofastidiosa (Nobu et al., 2016) y los nuevos filos de Bathyarchaeota (Evans et al., 2015) y Verstraetearchaeota (Vanwonterghem et al., 2016). Estos nuevos hallazgos ampliaron nuestros conocimientos sobre la diversidad de metanógenos potenciales, pero no oscurecieron la aplicabilidad del gen mcrA como marcador molecular para la gran mayoría de las comunidades metanógenas.
Hasta la fecha, se han detectado comunidades metanógenas en humedales, sedimentos, zonas de permafrost, arrozales, digestores, manantiales geotérmicos y respiraderos hidrotermales (Conrad, 2007; Thauer et al., 2008; Wagner y Liebner, 2009). Se ha descubierto que la estructura de la comunidad metanogénica está asociada con el pH ambiental, la temperatura, la salinidad, el nivel del agua subterránea y la dinámica de la vegetación a diferentes escalas espaciales y temporales (Megonigal et al., 2005; Milferstedt et al., 2010; Frank-Fahle et al., 2014; McCalley et al., 2014; Cui et al., 2015; Liebner et al., 2015). Por ejemplo, la metanogénesis acetoclástica suele verse obstaculizada por un pH bajo, ya que reduce la disociación del acetato (Megonigal et al., 2005; Kotsyurbenko et al., 2007). La vegetación puede suministrar carbono orgánico lábil y de alta calidad para alimentar a los metanógenos en forma de exudados de las raíces o de detritus, de modo que los exudados de las plantas suelen favorecer a los metanógenos acetoclásticos principalmente en los pantanos (Bridgham et al., 2013). El sulfato del agua de mar inhibe la producción de metano en los humedales de marea y, en consecuencia, la salinidad se ha utilizado como predictor general de las emisiones de metano (Holm et al., 2016). Un estudio sobre los sedimentos de los lagos tibetanos demostró que el aumento de la salinidad inhibe los metanógenos hidrogenotróficos pero potencia la metanogénesis acetoclástica (Liu et al., 2016). Estos estudios indicaron impulsores ambientales para las comunidades metanogénicas, pero se han centrado en hábitats únicos o en escalas espaciales limitadas.
La comprensión de la adaptación de los metanógenos a diferentes cambios ambientales, sin embargo, requiere una exploración sistemática y global de las correlaciones entre la composición de la comunidad microbiana y las condiciones ambientales (Lozupone y Knight, 2007). En la actualidad, sólo unos pocos estudios abordan la dispersión y el filtrado del hábitat de las comunidades metanógenas (Auguet et al., 2010; Barreto et al., 2014). Nuestra hipótesis es que los ensamblajes metanogénicos están influenciados principalmente por el filtrado del hábitat y que éste es impulsado por los controles ambientales globales. Teniendo en cuenta que las tasas de emisión de metano difieren en gran medida entre los ecosistemas naturales, la integración explícita de la composición, la diversidad y la biogeografía de los conjuntos metanogénicos en estos ecosistemas puede ser fundamental para determinar la respuesta de la producción de metano al cambio climático actual y futuro. Este meta-estudio se realiza para llenar los vacíos asociados con la biogeografía metanogénica, la diversidad y sus controles ambientales mediante el uso de datos de secuencias mcrA disponibles públicamente y la literatura complementada a través de los datos fisiológicos de los aislados metanogénicos.
Materiales y Métodos
Colección de datos
Recuperamos secuencias mcrA disponibles en GenBank (enero de 2016)2. Para cada resultado, se comprobó el artículo original y se analizaron las secuencias mcrA correspondientes mediante un script Perl personalizado. Como nos centramos en los entornos naturales, las secuencias mcrA metanogénicas se obtuvieron de hábitats naturales y se clasificaron como suelo, lago, estuario, sedimentos marinos e hidrotermales y volcanes de lodo. Se incluyeron cinco bibliotecas de secuenciación de próxima generación (NGS) además de las secuencias de las bibliotecas de clones. Las secuencias se descargaron sin tener en cuenta la abundancia relativa en el conjunto de datos original. Dado que las secuencias de las bibliotecas de clones cubren principalmente los filotipos abundantes, mientras que la NGS puede capturar una diversidad mucho más profunda, hicimos un compromiso para utilizar los datos de la NGS, pero mitigando un posible error debido a la diferente resolución de los métodos de secuenciación. Por lo tanto, sólo elegimos las secuencias representativas de OTUs abundantes con una abundancia relativa superior al 1%. Además, rechazamos aquellas secuencias NGS que no superaron la comprobación de traducción de secuencias de nucleótidos a proteínas o con una calidad baja (secuencias < 250 bps). Finalmente, construimos un conjunto de datos que contenía 4.466 secuencias únicas de mcrA de 94 sitios distribuidos globalmente (Figura 1 y Tablas Suplementarias S1, S2). Además, no sustrajimos del conjunto de datos las secuencias mcrA de posibles metanótrofos arqueológicos, que inevitablemente se detectaron en el estudio genómico (Conrad, 2007). Esta parte está fuera del enfoque de este estudio.
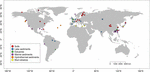
FIGURA 1. Ubicación de los sitios utilizados para este estudio. Los sitios se agruparon en seis categorías diferentes según sus hábitats y se etiquetaron con diferentes colores. Un resumen de los seis hábitats se da en las Tablas Suplementarias S1, S2.
Las coordenadas geográficas y los parámetros ambientales, incluyendo el pH, la salinidad, la elevación, la temperatura media anual del aire (MAAT) y la precipitación media anual (MAP) se extrajeron para cada sitio de investigación considerado en este estudio de las publicaciones correspondientes dado que los datos están disponibles (ver Tabla Suplementaria S1). Para dar cuenta de los parámetros ambientales faltantes a través de múltiples estudios, definimos cualitativamente algunas variables ambientales de acuerdo con las descripciones de los sitios en la literatura relevante, y luego convertimos estos datos de categoría en valores numéricos semimétricos, por ejemplo, definimos los sedimentos marinos, los sedimentos hidrotermales, el lodo volcánico y las muestras de sedimentos de lagos de soda como «salinos», el suelo y las muestras de sedimentos de lagos de agua dulce como «no salinos», y las muestras de manglares y estuarios como muestras «mixtas».
Procesamiento de secuencias crudas
El procesamiento de secuencias se implementó con la plataforma de software Mothur (Schloss et al, 2009). Las secuencias de diferentes bibliotecas se agruparon antes del procesamiento. Se descartaron las secuencias con una longitud inferior a 350 pb o con más de 8 bases ambiguas. Posteriormente, estas secuencias se alinearon contra un subconjunto prealineado de secuencias de mcrA, que se recuperaron de la base de datos FunGene en http://fungene.cme.msu.edu/ (Fish et al., 2013). Las secuencias quiméricas se identificaron con el software Mothur utilizando el método uchime (Edgar et al., 2011) con el propio conjunto de datos como referencia. A continuación, las secuencias nucleotídicas válidas del gen mcrA se utilizaron para calcular las distancias por pares no corregidas entre las secuencias de ADN alineadas y, además, se asignaron en unidades taxonómicas operativas (OTU) con un corte del 84% que corresponde al 97% del gen 16S rRNA (Yang et al., 2014). La abundancia de cada OTU de mcrA solo se contabilizó como presencia o ausencia. Aumentamos la precisión de la clasificación taxonómica de las OTUs considerando tanto las secuencias de nucleótidos como las de aminoácidos. A nivel de ADN, la identidad taxonómica fue asignada por la plataforma Mothur según una base de datos de referencia (Yang et al., 2014). A nivel de proteínas, las secuencias de proteínas alineadas se utilizaron para construir un árbol en ARB, y luego la asignación taxonómica se basó en la base de datos correspondiente. Si la asignación de una OTU era incoherente, se realizó un blasting manual tanto de la secuencia de nucleótidos como de la de proteínas en NCBI y se determinó la identidad taxonómica final teniendo en cuenta la cobertura de la consulta (>95%), la identidad (>84%) y el valor e (<1E-5). Para las secuencias de proteínas, el corte a nivel de género se refirió al umbral del 83,5% (Hunger et al., 2011).
Análisis ecológico y estadístico
El análisis estadístico se realizó mediante varios paquetes de R. Las ordenaciones del análisis de coordenadas principales (PCoA) se generaron a partir de las matrices de distancia de Jaccard construidas con el paquete vegano v2.2.0 (Oksanen et al., 2015). Se realizó un MANOVA permutacional (análisis multivariante de la varianza) para evaluar el origen de la variación en la matriz de Jaccard (McArdle y Anderson, 2001) en el vegano con 104 permutaciones. Las medidas de distancia de Jaccard se basan en la presencia/ausencia de la especie, lo que es más adecuado para nuestro conjunto de datos, ya que la mayoría de los estudios sólo proporcionaron las secuencias representativas, mientras que falta la información sobre las abundancias. Las frecuencias de incidencia taxonómica en los hábitats se visualizaron mediante gráficos de burbujas con el paquete ggplot2 (v1.0.0) (Wickham, 2009). El análisis de agrupación jerárquica de las comunidades del suelo no salino y de los sedimentos lacustres se realizó mediante la función de R ‘hclust’ (R Core Team, 2014). Los clústeres de comunidades obtenidos se describieron de acuerdo con el régimen de pH y temperatura de las muestras originales, ya que usando PCoA de antemano identificamos que ambos parámetros, pH y temperatura, influyen en la composición de la comunidad metanogénica en los suelos no salinos y en los sedimentos lacustres. La asociación de cada linaje metanogénico con cada uno de estos grupos se determinó mediante un análisis de especies indicadoras basado en la correlación (Dufrene y Legendre, 1997). Las especies indicadoras se definen aquí como aquellas que son abundantes en un tipo específico de hábitat (especificidad) y que se encuentran predominantemente en este tipo de hábitat (fidelidad). En este estudio, los taxones indicadores, similares al concepto de especies indicadoras, para los suelos no salinos y los sedimentos lacustres se eligieron según un valor indicador (valor IndVal) mediante el paquete R labdsv (Roberts, 2016) si la probabilidad de obtener un valor indicador tan alto como el observado a lo largo de las iteraciones especificadas es inferior a 0,05. Los índices Chao2 se calcularon para cada muestra utilizando el paquete vegano. La prueba de suma de rangos de Wilcoxon de los índices Chao2 entre hábitats se realizó mediante la función de R ‘wilcox.test’ (R Core Team, 2014). Para consolidar el impacto del filtrado del hábitat en los aislados de archaea metanogénica, se recuperaron las características fisiológicas y bioquímicas de los cultivos metanogénicos descritos en la3 ‘Methanogenic archaea database’ (Jabłoński et al., 2015). Entre ellos, los aislados con información de requerimiento óptimo de NaCl fueron filtrados, categorizados y trazados según su fuente de aislamiento.
Para examinar la influencia de la limitación de la dispersión en la estructura de la comunidad metanogénica, se realizó un análisis de regresión lineal basado en una matriz de distancia geográfica y una matriz de distancia de Jaccard de la comunidad mediante la función de R ‘lm’ (R Core Team, 2014). Realizamos las pruebas de Mantel y de Mantel parcial para evaluar los efectos de la limitación de la dispersión según las dos matrices de nuevo utilizando el paquete vegan en R (Oksanen et al., 2015). Además, se aplicó un análisis espacial multivariante (PCA espacial) a 16 muestras europeas de suelo y sedimentos lacustres basado en el índice I de Moran para explorar la estructura espacial de los metanógenos mediante la función «multispati» del paquete R ade4 (Dray y Dufour, 2007). Además, la agrupación de varianza mínima de Ward, basada en la matriz de distancia de Jaccard, se implementó en estas 16 muestras utilizando la función de R «hclust» (R Core Team, 2014) y proyectamos los resultados de la agrupación en un mapa geográfico. El shapefile europeo para la cartografía a nivel estatal está disponible en la base de datos GSHHG (v2.3.6)4. El mapa se generó utilizando QGIS v2.18.25.
Resultados
Biogeografía de Arqueas Metanogénicas en Ambientes Naturales
Se recuperaron las secuencias del gen mcrA de 94 ambientes naturales distribuidos globalmente. La ubicación y el tipo de ecosistema de cada uno de estos 94 sitios se representa en la Figura 1. Las frecuencias de incidencia (presencia/ausencia) de los linajes metanógenos se fusionaron según el tipo de ecosistema y se ilustran en la Figura 2. En resumen, Methanoregula es el taxón más frecuentemente observado en los suelos, junto con Methanobacterium, Methanosaeta, Methanocella, Methanomassiliicoccus y Methanosarcina. En los sedimentos de estuario, se detectaron comúnmente secuencias de Methanosaeta, Methanobacterium, Methanoregula y Methanoculleus. Además, en los sedimentos lacustres, se encontraron principalmente Methanoregula y Methanosaeta. En los sedimentos marinos, Methanoculleus y Methanosaeta son los linajes más comunes, seguidos de Methanolinea.
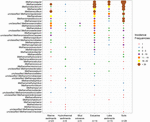
FIGURA 2. Gráfico de burbujas que muestra las frecuencias de incidencia de los linajes metanogénicos en diferentes ambientes naturales. El orden de clasificación a lo largo del eje vertical corresponde a las frecuencias de incidencia totales decrecientes de los linajes. La taxonomía se muestra para el nivel de género. Si no fue posible la asignación al nivel de género, se utilizó el siguiente nivel taxonómico asignable. Se indica el número de muestras (n) para cada hábitat.
Aunque se detectaron muchos taxones en diferentes ambientes, algunos siguen mostrando preferencias ambientales. Methanoregula, el taxón que aparece frecuentemente en ambientes no marinos y de transición (suelos, sedimentos lacustres y estuarios), está ausente de los hábitats marinos (sedimentos marinos, sedimentos hidrotermales y volcanes de lodo). Methanobacterium y Methanocella, que predominan en los ambientes no marinos y de transición, se encuentran raramente en los hábitats marinos. En cambio, Methanococcoides, como linaje predominante en sedimentos marinos, sedimentos hidrotermales y volcanes de lodo, apenas se observa en suelos y sedimentos lacustres. Además, Methanogenium y Methanolacinia sólo se observan en estuarios y sedimentos marinos, mientras que Methanospirillum y Methanosphaerula sólo se encuentran en ambientes terrestres. Además, algunos taxones específicos se encuentran exclusivamente en sedimentos hidrotermales, como Methanocaldococcus, Methanothermococcus, Methanopyrus, Methanotorris y Methanococcus. Aunque algunos linajes como Methanosaeta están presentes en la mayoría de los ambientes, ningún linaje puede considerarse como omnipresente.
La mayor riqueza de linajes se dio en los sedimentos de estuario que también albergan frecuencias de incidencia más uniformes de varios linajes. Por el contrario, los ecosistemas de volcanes de lodo e hidrotermales muestran una diversidad metanogénica relativamente baja. Los suelos y los sedimentos lacustres, similares a los de los estuarios, se caracterizan por la diversidad de los conjuntos metanogénicos.
Diversidad alfa de las comunidades metanogénicas en entornos naturales
La riqueza de las arqueas metanogénicas según el índice Chao2 varió en gran medida entre los distintos tipos de ecosistemas (Figura 3). Con el fin de comparar directamente las diversidades alfa y para obtener una compensación razonable entre las muestras, se realizó un submuestreo a 15 secuencias para cada sitio. El índice Chao2 muestra que los sedimentos de estuario abarcan la mayor riqueza de especies de metanógenos a lo largo de los seis tipos de ecosistemas (Tabla Suplementaria S3), lo que subraya los resultados del gráfico de burbujas (Figura 2). Los suelos y los sedimentos lacustres, que muestran una menor riqueza que las muestras del estuario, tienen índices Chao2 significativamente más altos que los sedimentos marinos y los sedimentos hidrotermales y sin diferencias significativas entre los sedimentos marinos y los sedimentos hidrotermales (Tabla Suplementaria S3).
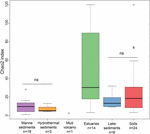
FIGURA 3. Diagrama de cajas de los índices Chao2 de los diferentes tipos de ecosistemas. El gráfico se basa en conjuntos de datos submuestreados que contienen 15 secuencias para cada sitio para hacer más robusta la comparación sobre las medidas de diversidad alfa. El número de muestras en cada hábitat se da como ‘n’ debajo de la etiqueta del hábitat. La «ns» indica que no hay significación estadística en la prueba de Wilcoxon. El resultado estadístico de la diversidad alfa a nivel de OTU se da en la Tabla Suplementaria S3.
Control global de las comunidades metanogénicas en ambientes naturales
Las 94 comunidades metanogénicas distribuidas globalmente se agruparon en un gráfico de ordenación aplicando el PCoA basado en la matriz de distancia de Jaccard. Según el análisis PCoA, los ejes primero y segundo explican conjuntamente el 16,3% de la varianza total. Por lo tanto, las variaciones entre las muestras pueden explicarse en gran medida por la salinidad (Figura 4). Dado que en algunos casos no se disponía de datos iniciales sobre las concentraciones de sal, asignamos cualitativamente estas muestras como salinas, mixtas (intermedias) y no salinas, tal como se ha descrito anteriormente. Las muestras salinas y no salinas se separan efectivamente a lo largo del primer eje. Las muestras mixtas se agrupan en general entre las muestras salinas y las no salinas. El MANOVA permutacional basado en la matriz de distancia de Jaccard también sugiere que la salinidad es el principal factor abiótico que controla la distribución de las comunidades metanogénicas globales (R2 = 0,099, P < 0,001) (Tabla 1).
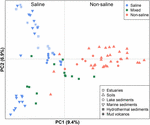
FIGURA 4. Ordenación del análisis de coordenadas principales (PCoA) basada en la matriz de distancia de Jaccard de las bibliotecas de secuencias del gen mcrA metanogénico comparando 94 muestras. El PCoA está coloreado según la salinidad: Los símbolos rojos indican ambientes no salinos, los azules ambientes salinos y los verdes ambientes intermedios. Diferentes símbolos representan diferentes ambientes. El porcentaje de la variación explicada por las coordenadas principales trazadas se indica en los ejes.

TABLA 1. Análisis MANOVA permutacional sobre la matriz de distancia de Jaccard de todas las muestras de los seis hábitats para comprobar la asociación de la varianza de la comunidad con diferentes variables ambientales.
Además, se comprobó la posible relación entre la fuente de aislamiento de los cultivos puros metanogénicos y la concentración óptima de NaCl para su crecimiento. La concentración óptima de NaCl de los cultivos puros metanogénicos demostró un descenso de los ecosistemas marinos a los estuarios, los sedimentos lacustres y el suelo (Figura Suplementaria S2). Unos pocos aislados atípicos proceden de sedimentos de lagos de sosa o de suelos hipersalinos.
Controles ambientales y taxones indicadores metanogénicos en suelos no salinos y sedimentos lacustres
A escala global, las comunidades metanogénicas de suelos no salinos y sedimentos lacustres se agrupan estrechamente (Figura 4), por lo que analizamos más a fondo los controles ambientales de las comunidades metanogénicas de estos dos hábitats, que representan 33 sitios de estudio en total. El análisis de conglomerados basado en la comunidad para estos dos tipos de hábitats reveló cuatro conglomerados basados en la matriz de distancia de Jaccard (Figura Suplementaria S3). El MANOVA permutacional sugiere que tanto el pH (R2 = 0,099, P < 0,001) como la temperatura (R2 = 0,069, P < 0,001) influyen en la β-diversidad metanogénica en los suelos no salinos y en los sedimentos lacustres (Tabla 2). En consecuencia, asignamos los cuatro clusters al pH y al MAAT del sitio de muestreo inicial y obtuvimos subgrupos ampliamente consistentes con la agrupación de la comunidad (Figuras 5C,D). La combinación de las características ambientales y estos cuatro clusters de la comunidad nos permite definir estos cuatro subgrupos como grupo neutro y frío 1, grupo ácido y frío 2, grupo ácido y moderado 3, y grupo neutro y cálido 4 (Figura 5). Una nueva ordenación del PCoA basada en la matriz de disimilitud de Jaccard sugiere que, a lo largo del eje PC1, la mayoría de las muestras del grupo 2 y del grupo 3 proceden de suelos ácidos y sedimentos lacustres, mientras que el grupo 1 y el grupo 4 proceden principalmente de entornos neutros (Figura 5A). Además, las muestras de sitios moderados (grupo 3) se separaron de las de sitios cálidos y fríos a lo largo de PC2, mientras que las muestras de los ambientes cálidos (grupo 4) se separaron de las otras muestras a lo largo de PC3 (Figura 5B). De este modo, los tres primeros ejes de la ordenación PCoA explican el 38,8% de la variación total.

Tabla 2. MANOVA permutacional basado en una matriz de distancia de Jaccard de muestras de suelo no salino y sedimento lacustre para comprobar la asociación de la varianza de la comunidad con diferentes variables ambientales.
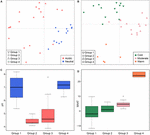
FIGURA 5. Gráfico de análisis de coordenadas principales basado en una matriz de distancia de Jaccard para 33 suelos no salinos y sedimentos lacustres. El subgrupo (A) muestra PC1 y PC2 y los símbolos están coloreados por el pH, y el subgrupo (B) muestra PC2 y PC3 y los símbolos están coloreados por la temperatura. Los tres primeros componentes explican el 17,1, el 11,7 y el 10% de la varianza. Los gráficos de caja muestran el pH (C) y la MAAT (temperatura media anual del aire) (D) de cuatro subgrupos identificados. El color de la caja en la figura C corresponde a la categoría de pH en (A). Los colores de los boxplots muestran la significación estadística basada en una prueba de Wilcoxon por pares (P < 0,05), en la que las muestras con el mismo color no difieren significativamente entre sí. Asimismo, el color en (D) sigue la agrupación de temperaturas en (B). Los subgrupos se refieren al análisis de cluster jerárquico de las similitudes de la comunidad.
Examinamos la ocurrencia de linajes metanogénicos en cada subgrupo basado en las frecuencias de incidencia (Figura 6). Methanoregula prevalece en todos los tipos de hábitats no salinos. Además de Methanoregula, el subgrupo neutro y frío (grupo 1) muestra una alta abundancia de Methanosaeta, Methanobacterium y Methanosarcina. El grupo ácido y frío 2 está representado por Methanobacterium, Methanocella y Methanosarcina, mientras que Methanosaeta está ausente. Methanocella y Methanosaeta son comunes en el grupo ácido y moderado 3. En el grupo neutro y cálido 4, Methanolinea y Methanosaeta son miembros importantes. Este grupo es el único en el que se identificó Methanoculleus.

FIGURA 6. Frecuencias de incidencia de los linajes metanogénicos dentro de los cuatro grupos definidos de comunidades metanogénicas en suelos y sedimentos lacustres. El eje vertical está dispuesto en orden alfabético. La longitud de las barras corresponde a las frecuencias medias de incidencia de cada linaje dentro del grupo correspondiente. Las barras de error representan la desviación estándar de un determinado taxón sobre diferentes muestras de ese grupo. Los asteriscos muestran los taxones especializados con un valor P < 0,05. La taxonomía se muestra para el nivel de género. Si no fue posible la asignación al nivel de género, se utilizó el siguiente nivel taxonómico asignable. La descripción de los cuatro grupos se muestra en la Figura 5.
Para los cuatro grupos, los taxones que tienen una alta incidencia son Methanoregula, Methanobacterium, Methanosarcina, Methanosaeta, Methanomassiliicoccus y Methanocella. Los taxones especializados, que están significativamente más representados en la mayoría de los sitios dentro de un grupo determinado, se detectaron según el análisis de especies indicadoras descrito anteriormente. En total, seis de los 31 taxones mostraron un valor indicador significativo (P < 0,05) (marcados con asterisco en la Figura 6). El grupo 1 (neutro y frío) mostró el mayor número de especialistas con linajes de Methanosaeta, Methanolobus y Methanomethylovorans. Methanobacterium fue un taxón especialista en el grupo 2 (frío y ácido), mientras que Methanolinea se identificó como especialista en el grupo 4 (neutro y cálido), pero apenas se observó en otros grupos. Además, Methanoregula está ampliamente representada en el grupo 3, ácido y moderado.
Limitación de la dispersión
Un análisis de regresión lineal (R2= 0,05, P < 0,001) indicó una débil correlación entre la distancia geográfica y la estructura de la comunidad metanogénica en el conjunto de datos global. Al mismo tiempo, una prueba de Mantel mostró que las variables ambientales tienen una mayor correlación con la estructura de la comunidad que las distancias geográficas (ver Tabla 3). Esta tendencia también se confirma mediante una prueba de Mantel parcial que controla los efectos de autocorrelación. El trazado de la distancia geográfica frente a la similitud de la comunidad de Jaccard no muestra ninguna tendencia lineal clara, sino patrones que resultan principalmente de la distribución global de los puntos de muestreo (véase la figura suplementaria S4).

TABLA 3. Análisis de Mantel y pruebas parciales de Mantel para la determinación de la influencia de las variables ambientales y la distancia geográfica en la distribución microbiana para el conjunto de datos global y una submuestra de 16 muestras europeas.
Con el fin de analizar más a fondo la influencia de la dispersión, limitamos nuestro análisis a Europa, que fue muestreada con mayor densidad y uniformidad. Las pruebas de Mantel y las pruebas parciales de Mantel en este subconjunto reprodujeron la tendencia de que los datos de la comunidad están más correlacionados con las variables ambientales que con las distancias geográficas (véase la Tabla 3). La prueba de Mantel parcial que controla las variables ambientales no pudo detectar ninguna correlación estadísticamente significativa entre la comunidad microbiana y la distancia geográfica. Un análisis PCA espacial en estos 16 sitios europeos implica una estructura espacial de la comunidad metanogénica (el 23,7% de la varianza total fue explicada por esta estructura) que corresponde a una autocorrelación espacial positiva de los sitios como indica el índice I de Moran (I de Moran = 0,4018). Sólo el primer valor propio era estable y correspondía a una separación de las muestras entre Europa Central y los Estados Bálticos (Figura suplementaria S5). Sin embargo, el pequeño conjunto de datos complica una asignación sólida de esta estructura espacial observada a variables geográficas, ambientales o a ambas. Por lo tanto, llevamos a cabo un análisis de conglomerados en las comunidades metanogénicas como se ha descrito anteriormente y revelamos tres grupos (Figura 7A) que proyectamos en un mapa geográfico (Figura 7B). La agrupación no reprodujo la separación del PCA espacial a lo largo del Mar Báltico. En consecuencia, algunos sitios que están geográficamente muy cerca unos de otros exhiben estructuras comunitarias metanogénicas disímiles y se reúnen con diferentes grupos. Por otro lado, algunos sitios geográficamente muy distantes muestran composiciones comunitarias muy similares y se agrupan juntos (Figura 7B). La dispersión regional de los grupos en Europa no parece corresponder o estar limitada por una estructura geográfica.
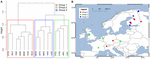
FIGURA 7. Análisis jerárquico de conglomerados de la matriz de distancia de Jaccard entre 16 muestras europeas de suelos y sedimentos lacustres. Para el análisis se utilizó el método de clustering de Ward. Los sitios se dividieron en tres grupos coloreados por diferentes rectángulos en el dendrograma de agrupación (A) y se proyectaron en un mapa europeo (B).
Discusión
La identificación y aplicación de conceptos de biogeografía en las comunidades microbianas es de gran interés en la ecología microbiana. Se cree que la biogeografía microbiana se rige por la interacción evolutiva y ecológica de cuatro procesos principales: filtrado del hábitat, dispersión, deriva y mutación (Hanson et al., 2012). Aunque la influencia de la deriva y la mutación está más allá del poder de interpretación de este estudio, demostramos que existen patrones globales de comunidades metanogénicas en entornos naturales. Este estudio demuestra un patrón biogeográfico global de las comunidades metanogénicas que está más asociado al filtrado del hábitat que a la dispersión geográfica. Las comunidades metanogénicas de los sedimentos de los lagos de sosa, por ejemplo, se agrupan estrechamente con las muestras marinas geográficamente distantes (Figura 4) y se dan comunidades metanogénicas muy similares en los sedimentos de los suelos y lagos europeos a pesar de estar situados a gran distancia unos de otros (Figura 7). En general, nuestros intentos de revelar la posible limitación de la dispersión revelaron una débil influencia de la ubicación geográfica en la estructura de la comunidad metanogénica que contrasta con una clara influencia de las condiciones ambientales. Una exclusión concluyente de los efectos espaciales sobre las comunidades microbianas no es posible con los puntos de datos disponibles. Los puntos de muestreo se centran en ciertos hábitats y/o áreas mientras que el número total de muestras es bajo. Si hay un efecto, suponemos que los efectos espaciales se producen sólo a escala regional o local. Se ha informado de limitaciones de dispersión local para las arqueas hipertermófilas causadas por barreras geográficas (Whitaker et al., 2003), bacterias oxidantes del amoníaco en marismas locales, pero no a escala regional y continental (Martiny et al., 2011), microorganismos en sedimentos de aguas profundas junto con entornos ambientales (Schauer et al., 2009), y para las bacterias de un gran conjunto de entornos de nieve heterogéneos causados principalmente por la disponibilidad de carbono alóctono (Lutz et al., 2016). Una influencia menor de la limitación de la dispersión en los metanógenos en entornos naturales significa que los metanógenos pueden distribuirse aleatoriamente en el espacio, son seleccionados con éxito por el entorno local si se cumplen sus requisitos fisiológicos y pueden establecer comunidades estables (Martiny et al., 2006; von Mering et al., 2007).
El concepto de filtrado del hábitat implica que las especies con requisitos ecológicos similares deben co-ocurrir más a menudo de lo esperado por el azar (Weiher y Keddy, 2001; Cornwell et al., 2006; Ulrich et al., 2010). Nuestros resultados muestran que existen grandes diferencias en la composición de la comunidad metanogénica entre los ecosistemas marinos y los de humedales y lagos, mientras que los estuarios se agrupan en el medio. También se informó de patrones biogeográficos basados en un conjunto de estudios de genes en muestras ambientales para las bacterias generales (Nemergut et al., 2011), el microbioma de las filtraciones de metano (Ruff et al., 2015), las arqueas que oxidan el amoníaco (Cao et al., 2013), las bacterias marinas pelágicas y bentónicas (Zinger et al., 2011) y los microorganismos que reciclan el nitrógeno (Church et al., 2008). El filtrado de hábitats fue reportado específicamente para arqueas no cultivadas (Auguet et al., 2010), comunidades bacterianas enteras en diversos ambientes (von Mering et al., 2007; Chaffron et al., 2010) o en turberas sudamericanas a escala regional (Oloo et al., 2016), así como grupos bacterianos específicos como las bacterias oxidantes de metano (Knief, 2015) y las fijadoras de nitrógeno (Nelson et al., 2016).
Nuestros resultados indican que, a escala global, la salinidad regula sustancialmente la composición de la comunidad metanogénica y determina grandes diferencias entre los conjuntos metanogénicos marinos y terrestres. También los metanógenos de los sedimentos de los lagos de sosa se agrupan con los de los sedimentos marinos (Figura 4), lo que pone de manifiesto la influencia global de la salinidad. Este resultado concuerda con otros estudios basados en el gen 16S rRNA que revelan que la salinidad es un factor primario que da forma a los patrones globales de las comunidades bacterianas y arqueas globales (Lozupone y Knight, 2007; Auguet et al., 2010; Caporaso et al., 2011; Cao et al., 2013). En estudios anteriores también se observó una baja influencia de la separación geográfica pero un fuerte impacto de la salinidad en las comunidades microbianas generales (Logares et al., 2013; Yang et al., 2016). En consecuencia, la salinidad determina en gran medida qué linajes pueden sobrevivir. En varios hábitats la actividad de producción de metano se correlacionó negativamente con la salinidad (Bartlett et al., 1987; Potter et al., 2009; Poffenbarger et al., 2011). Por lo tanto, se sugiere que la inhibición de la producción de metano a través de la salinidad coincide con un tamaño reducido de la población metanogénica (Pattnaik et al., 2000). El efecto de la salinidad en la metanogénesis hidrogenotrófica, acetotrófica y metilotrófica depende, por tanto, del nivel de salinidad y es diferente para las distintas vías de metanogénesis (Liu et al., 2016). Actualmente, no existe un mecanismo claro para explicar el impacto de la salinidad en la estructura de la comunidad, pero varias hipótesis pueden servir como posible explicación. Fisiológicamente, la salinidad influye en la osmolaridad externa e interna de las células. Las células metanogénicas no salinas han desarrollado adaptaciones fisiológicas para contrarrestar la presión de turgencia interna, mientras que las células adaptadas a la sal han perdido tal característica (Zinder, 1993). Además, el aumento de la salinidad puede inducir a los metanógenos a sintetizar o tomar una mayor proporción de solutos compatibles con un importante coste energético y, por tanto, metabólico (McGenity, 2010). El rasgo de la tolerancia a la sal se manifiesta incluso en la concentración óptima de NaCl para el crecimiento de los cultivos puros de metanógenos, ya que encontramos que los aislados de sedimentos marinos y sedimentos hidrotermales tienen una concentración óptima de NaCl significativamente mayor que los de los suelos (Figura Suplementaria S2).
En los ecosistemas terrestres no salinos, concretamente en suelos y sedimentos lacustres, la composición de la comunidad metanogénica está controlada por la combinación de temperatura y pH. En consecuencia, los metanógenos de estos ambientes podrían clasificarse en cuatro grupos (Figura 6). A diferencia de los ecosistemas marinos, los ecosistemas terrestres no salinos muestran una gran variabilidad natural tanto de pH como de temperatura. La temperatura puede afectar no sólo a la vía metanogénica sino también a las propias poblaciones metanogénicas (Conrad, 2007; Rooney-Varga et al., 2007). La producción de metano puede aumentar considerablemente si la temperatura se eleva como consecuencia de los pasos sensibles a la temperatura durante la fermentación y la acetogénesis (Megonigal et al., 2005; Kotsyurbenko et al., 2007). Además, un pH bajo puede limitar sustancialmente la disponibilidad de acetato al impedir que éste se disocie y, por tanto, afectar negativamente a la metanogénesis acetoclástica (Fukuzaki et al., 1990; Bridgham et al., 2013). Esta podría ser una posible razón por la que Methanosaeta estuvo ausente en el grupo2, mientras que Methanosarcina puede cambiar entre diferentes fuentes y no se vio sustancialmente influenciada. Además, el pH puede regular la eficiencia de la producción de metano y las vías metanogénicas de las turberas ombrotróficas a las minerotróficas, a través de la inhibición directa de ambas vías de metanogénesis e indirectamente a través de sus efectos en la fermentación (Ye et al., 2012). Por lo tanto, tanto la temperatura como el pH pueden regular directa o indirectamente los pasos metabólicos asociados a la metanogénesis y a la fermentación ascendente, que proporciona el sustrato para los metanógenos.
La metanoregula es ubicua y muy abundante en los cuatro grupos de hábitats terrestres (Figura 6), pero está prácticamente ausente en el sistema marino y, por lo tanto, puede resultar un indicador de la influencia del agua dulce en el ámbito marino. Su relevancia global se informó recientemente en otro lugar (Yang et al., 2017). A pesar de su distribución ubicua en suelos y sedimentos lacustres, Methanoregula se presenta como un linaje indicador en hábitats ácidos con temperaturas moderadas. Además, (1) Methanolinea parece haberse adaptado especialmente a los entornos terrestres cálidos y neutros, (2) Methanobacterium a los entornos fríos y ácidos, y (3) Methanosaeta a los entornos de pH neutro, lo que coincide con otros estudios (Rosenberg et al., 2014) y subraya la solidez de nuestro enfoque. En general, las condiciones geoquímicas que rodean a las comunidades metanogénicas conducirán a la diferenciación de nichos. Dado que la clasificación de nichos tiende a dejar a los especialistas adaptativos (Langenheder y Székely, 2011), la selección ambiental progresiva a largo plazo generó una variedad de nichos que fueron ocupados por una serie de especialistas endémicos del hábitat, que pueden estar menos representados o ausentes en otras condiciones ambientales diferentes. La comunidad también está formada por factores bióticos, como las interacciones ecológicas, la dinámica, la competencia y la simbiosis. A pesar de esos factores biológicos, von Mering et al. (2007) descubrieron que las preferencias de hábitat suelen ser notablemente estables a lo largo del tiempo y que la composición taxonómica distintiva de las comunidades ambientales, a su vez, puede ser un importante indicador de su ecología y función.
En consonancia con la preferencia de hábitat de las arqueas metanogénicas, parece que cepas metanogénicas estrechamente relacionadas se aislaron a menudo de entornos comparables. Por ejemplo, Methanoregulaceae parece ser bastante diversa en entornos naturales (Yang et al., 2017) y las cepas neutrales pueden resistir el cultivo hasta ahora. Las cepas de Methanoregula descritas actualmente se obtienen de entornos ligeramente acidófilos, mientras que los dos representantes de Methanolinea proceden de hábitats relativamente cálidos, como los lodos de digestores y el suelo de los arrozales, respectivamente (Rosenberg et al., 2014). Aunque las cepas de Methanobacterium fueron aisladas de diversos ambientes, aproximadamente la mitad de los aislados existentes de este género exhiben óptimos de pH ligeramente inferiores a 7. Esto significa que los linajes indicadores, que fueron identificados en base a las secuencias ambientales, podrían reflejar las diferenciaciones de fisiología y fuentes de los cultivares metanogénicos existentes. Por ejemplo, la salinidad del hábitat, como propiedad general del mismo, puede exponer progresivamente a los organismos a una fuerte selección ambiental y filtrar el ensamblaje de un nuevo conjunto de especies que se adaptan mejor a la salinidad ambiental (Logares et al., 2013).
La conservación y gestión de la biodiversidad es un reto primordial de nuestra sociedad actual. Aquí mostramos que las arqueas metanogénicas de los ambientes naturales son más diversas en los sedimentos de los estuarios. Los estuarios son zonas de transición entre los ecosistemas marinos y terrestres. Esto permite dos procesos principales que pueden contribuir a la riqueza de especies. En primer lugar, los microbios del mar y de la tierra se mezclan en los estuarios y acaban englobando una gran diversidad general (McLusky y Elliott, 2004). Por ejemplo, se ha observado una gran diversidad de bacterias de estuario, arqueas, hongos e incluso bacterias específicas que desempeñan funciones únicas (Cunliffe et al., 2008; Mosier y Francis, 2008; Crump et al., 2012). Otro aspecto es el alto nivel de nutrientes debido a los aportes terrestres y de las mareas para que los organismos del estuario se alimenten (McLusky y Elliott, 2004; Statham, 2012). En este contexto, los ambientes estuariales son importantes para recuperar la novedad genérica de los metanógenos. Hasta ahora, los efectos de la diversidad de especies en los procesos de los ecosistemas han atraído importantes esfuerzos de investigación. El vínculo entre la biodiversidad y la función de los ecosistemas sigue siendo objeto de debate y sigue siendo difícil de alcanzar para las comunidades microbianas (Loreau et al., 2001; Tilman et al., 2014). Aunque los suelos y los sedimentos lacustres son fuentes primarias de metano y también hábitats con una alta diversidad metanogénica, proponemos que la riqueza de especies no es un proxy apropiado del potencial de producción de metano y de las emisiones de metano del ecosistema; más bien parece reflejar la heterogeneidad y la historia del entorno. La clasificación de los ambientes según su riqueza de especies no significa necesariamente el potencial de las tasas de emisión de metano, que son más altas en los suelos y los lagos y comparativamente menores en los estuarios (Figura 3 y Figura Suplementaria S1).
Por último, la falta de información ambiental en las bases de datos públicas puede haber obstaculizado una interpretación completa sobre los factores ambientales observados aquí. Aunque hay cada vez más datos de secuenciación sobre metanógenos en la literatura y en las bases de datos públicas, las variables abióticas relacionadas proporcionadas son a menudo inconsistentes y escasas. La limitada cantidad de información consistente para las variables ambientales restringe la aplicación de análisis estadísticos multivariados. Si se recuerda que los factores abióticos en este estudio sólo pueden explicar una fracción limitada de las varianzas de la comunidad, se sugiere que faltan otras variables explicativas. De especial importancia podrían ser las concentraciones y la disponibilidad de sustratos metanogénicos como el acetato, el hidrógeno y las metilaminas. No obstante, es posible que los parámetros abióticos nunca sean suficientes para explicar completamente lo que estructura los conjuntos metanogénicos, simplemente porque los hábitats tienen historias diferentes y sólo pueden estudiarse localmente. Además, entre los datos disponibles de mcrA las vastas zonas del subártico y el ártico ruso y canadiense están poco representadas. Una mejor cobertura geográfica y una distribución uniforme del conjunto de datos de genes mcrA mejoraría una evaluación de las comunidades metanogénicas a escala global.
Contribuciones de los autores
SL y XW diseñaron el estudio. XW y SY recogieron y analizaron los datos. XW, SY y FH realizaron el análisis estadístico. XW, MW, y SY hicieron la corrección filogenética. XW, SY, FH, MW, DW y SL interpretaron los resultados y escribieron el artículo. Todos los autores contribuyeron a las discusiones y revisaron el manuscrito.
Declaración de conflicto de intereses
Los autores declaran que la investigación se llevó a cabo en ausencia de cualquier relación comercial o financiera que pudiera interpretarse como un potencial conflicto de intereses.
Agradecimientos
Se agradece el apoyo financiero a XW (subvención nº 201408620031 a XW) proporcionado por el Consejo de Becas de China (CSC). Este estudio fue apoyado por la Helmholtz Gemeinschaft (HGF) mediante la financiación del Grupo de Jóvenes Investigadores Helmholtz de SL (VH-NG-919).
Material suplementario
El material suplementario para este artículo se puede encontrar en línea en: https://www.frontiersin.org/article/10.3389/fmicb.2017.01339/full#supplementary-material
Notas al pie
- ^ http://metanogen.biotech.uni.wroc.pl/
- ^ http://www.ncbi.nlm.nih.gov/
- ^ http://metanogen.biotech.uni.wroc.pl/
- ^ https://www.ngdc.noaa.gov/mgg/shorelines/gshhs.html
- ^ http://qgis.osgeo.org
Auguet, J. C., Barberan, A., y Casamayor, E. O. (2010). Global ecological patterns in uncultured Archaea. ISME J. 4, 182-190. doi: 10.1038/ismej.2009.109
PubMed Abstract | CrossRef Full Text | Google Scholar
Bange, H. W., Bartell, U. H., Rapsomanikis, S., and Andreae, M. O. (1994). Methane in the Baltic and North Seas and a reassessment of the marine emissions of methane. Glob. Biogeochem. Cycles 8, 465-480. doi: 10.1029/94GB02181
CrossRef Full Text | Google Scholar
Barreto, D. P., Conrad, R., Klose, M., Claus, P., and Enrich-Prast, A. (2014). Relaciones de distancia-decadencia y taxa-área para bacterias, arqueas y arqueas metanogénicas en un sedimento de lago tropical. PLoS ONE 9:e110128. doi: 10.1371/journal.pone.0110128
PubMed Abstract | CrossRef Full Text | Google Scholar
Bartlett, K. B., Bartlett, D. S., Harriss, R. C., y Sebacher, D. I. (1987). Methane emissions along a salt marsh salinity gradient. Biogeochemistry 4, 183-202. doi: 10.1007/BF02187365
CrossRef Full Text | Google Scholar
Borrel, G., O’Toole, P. W., Harris, H. M. B., Peyret, P., Brugere, J. F., and Gribaldo, S. (2013). Los datos filogenómicos apoyan un séptimo orden de metanógenos metilotróficos y proporcionan información sobre la evolución de la metanogénesis. Genome Biol. Evol. 5, 1769-1780. doi: 10.1093/Gbe/Evt128
PubMed Abstract | CrossRef Full Text | Google Scholar
Bousquet, P., Ciais, P., Miller, J., Dlugokencky, E., Hauglustaine, D., Prigent, C., et al. (2006). Contribución de las fuentes antropogénicas y naturales a la variabilidad del metano atmosférico. Nature 443, 439-443. doi: 10.1038/nature05132
PubMed Abstract | CrossRef Full Text | Google Scholar
Bridgham, S. D., Cadillo-Quiroz, H., Keller, J. K., and Zhuang, Q. (2013). Methane emissions from wetlands: biogeochemical, microbial, and modeling perspectives from local to global scales. Glob. Chang. Biol. 19, 1325-1346. doi: 10.1111/gcb.12131
PubMed Abstract | CrossRef Full Text | Google Scholar
Cao, H., Auguet, J.-C., y Gu, J.-D. (2013). Global ecological pattern of ammonia-oxidizing archaea. PLoS ONE 8:e52853. doi: 10.1371/journal.pone.0052853
PubMed Abstract | CrossRef Full Text | Google Scholar
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516-4522. doi: 10.1073/pnas.1000080107
PubMed Abstract | CrossRef Full Text | Google Scholar
Chaffron, S., Rehrauer, H., Pernthaler, J., and von Mering, C. (2010). A global network of coexisting microbes from environmental and whole-genome sequence data. Genome Res. 20, 947-959. doi: 10.1101/gr.104521.109
PubMed Abstract | CrossRef Full Text | Google Scholar
Church, M. J., Bjorkman, K. M., Karl, D. M., Saito, M. A., and Zehr, J. P. (2008). Regional distributions of nitrogen-fixing bacteria in the Pacific Ocean. Limnol. Oceanogr. 53, 63-77. doi: 10.4319/lo.2008.53.1.0063
CrossRef Full Text | Google Scholar
Conrad, R. (2007). «Microbial ecology of methanogens and methanotrophs», en Advances in Agronomy, ed. L. S. Donald (Cambridge, MA: Academic Press), 1-63.
Google Scholar
Cornwell, W. K., Schwilk, D. W., y Ackerly, D. D. (2006). Una prueba basada en los rasgos para el filtrado del hábitat: el volumen del casco convexo. Ecology 87, 1465-1471. doi: 10.1890/0012-9658(2006)872.0.CO;2
PubMed Abstract | CrossRef Full Text | Google Scholar
Crump, B. C., Amaral-Zettler, L. A., and Kling, G. W. (2012). La diversidad microbiana en las aguas dulces árticas está estructurada por la inoculación de microbios de los suelos. ISME J. 6, 1629-1639. doi: 10.1038/ismej.2012.9
PubMed Abstract | CrossRef Full Text | Google Scholar
Cui, M., Ma, A., Qi, H., Zhuang, X., Zhuang, G., and Zhao, G. (2015). Una temperatura más cálida acelera las emisiones de metano del humedal de Zoige en la meseta tibetana sin cambiar la composición de la comunidad metanogénica. Sci. Rep. 5:11616. doi: 10.1038/srep11616
PubMed Abstract | CrossRef Full Text | Google Scholar
Cunliffe, M., Schafer, H., Harrison, E., Cleave, S., Upstill-Goddard, R., and Murrell, J. C. (2008). Phylogenetic and functional gene analysis of the bacterial and archaeal communities associated with the surface microlayer of an estuary. ISME J. 2, 776-789. doi: 10.1038/ismej.2008.28
PubMed Abstract | CrossRef Full Text | Google Scholar
Dray, S., and Dufour, A. B. (2007). El paquete ade4: implementación del diagrama de dualidad para los ecólogos. J. Stat. Softw. 22, 1-20. doi: 10.18637/jss.v022.i04
CrossRef Full Text | Google Scholar
Dufrene, M., y Legendre, P. (1997). Ensamblajes de especies y especies indicadoras: la necesidad de un enfoque asimétrico flexible. Ecol. Monogr. 67, 345-366. doi: 10.2307/2963459
CrossRef Full Text | Google Scholar
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME mejora la sensibilidad y la velocidad de detección de quimeras. Bioinformatics 27, 2194-2200. doi: 10.1093/bioinformatics/btr381
PubMed Abstract | CrossRef Full Text | Google Scholar
Evans, P. N., Parks, D. H., Chadwick, G. L., Robbins, S. J., Orphan, V. J., Golding, S. D., et al. (2015). Methane metabolism in the archaeal phylum Bathyarchaeota revealed by genome-centric metagenomics. Science 350, 434-438. doi: 10.1126/science.aac7745
PubMed Abstract | CrossRef Full Text | Google Scholar
Fish, J. A., Chai, B., Wang, Q., Sun, Y., Brown, C. T., Tiedje, J. M., et al. (2013). FunGene: la tubería y el repositorio de genes funcionales. Front. Microbiol. 4:291. doi: 10.3389/fmicb.2013.00291
PubMed Abstract | CrossRef Full Text | Google Scholar
Frank-Fahle, B. A., Yergeau, E., Greer, C. W., Lantuit, H., and Wagner, D. (2014). Microbial functional potential and community composition in permafrost-affected soils of the NW Canadian Arctic. PLoS ONE 9:e84761. doi: 10.1371/journal.pone.0084761.g001
PubMed Abstract | CrossRef Full Text | Google Scholar
Fukuzaki, S., Nishio, N., y Nagai, S. (1990). Kinetics of the methanogenic fermentation of acetate. Appl. Environ. Microbiol. 56, 3158-3163.
Google Scholar
Hanson, C. A., Fuhrman, J. A., Horner-Devine, M. C., y Martiny, J. B. (2012). Más allá de los patrones biogeográficos: procesos que dan forma al paisaje microbiano. Nat. Rev. Microbiol. 10, 497-506. doi: 10.1038/Nrmicro2795
PubMed Abstract | CrossRef Full Text | Google Scholar
Holm, G. O., Perez, B. C., McWhorter, D. E., Krauss, K. W., Johnson, D. J., Raynie, R. C., et al. (2016). Flujos de metano a nivel de ecosistema desde las marismas de agua dulce y salobre del delta del río Mississippi: implicaciones para los proyectos de carbono en humedales costeros. Wetlands 36, 401-413. doi: 10.1007/s13157-016-0746-7
CrossRef Full Text | Google Scholar
Hunger, S., Schmidt, O., Hilgarth, M., Horn, M. A., Kolb, S., Conrad, R., et al. (2011). Competing formate- and carbon dioxide-utilizing prokaryotes in anoxic methane-emitting fen soil. Appl. Environ. Microbiol. 77, 3773-3785. doi: 10.1128/AEM.00282-11
PubMed Abstract | CrossRef Full Text | Google Scholar
IPCC (2007). «Climate Change 2007: the Physical Science Basis», en Proceedings of the Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, eds D. Qin, S. Solomon, M. Manning, Z. Chen, M. Marquis, K. B. Averyt, et al. (Cambridge: Cambridge University Press).
Google Scholar
Jabłoński, S., Rodowicz, P., y Łukaszewicz, M. (2015). Base de datos de arqueas metanogénicas con características fisiológicas y bioquímicas. Int. J. Syst. Evol. Microbiol. 65, 1360-1368. doi: 10.1099/ijs.0.000065
PubMed Abstract | CrossRef Full Text | Google Scholar
Kirschke, S., Bousquet, P., Ciais, P., Saunois, M., Canadell, J. G., Dlugokencky, E. J., et al. (2013). Tres décadas de fuentes y sumideros globales de metano. Nat. Geosci. 6, 813-823. doi: 10.1038/Ngeo1955
CrossRef Full Text | Google Scholar
Knief, C. (2015). Diversidad y preferencias de hábitat de bacterias metanotróficas aeróbicas cultivadas y no cultivadas evaluadas en base a pmoA como marcador molecular. Front. Microbiol. 6:1346. doi: 10.3389/fmicb.2015.01346
PubMed Abstract | CrossRef Full Text | Google Scholar
Kotsyurbenko, O., Friedrich, M., Simankova, M., Nozhevnikova, A., Golyshin, P., Timmis, K., et al. (2007). Shift from acetoclastic to H2-dependent methanogenesis in a West Siberian peat bog at low pH values and isolation of an acidophilic Methanobacterium strain. Appl. Environ. Microbiol. 73, 2344-2348. doi: 10.1128/AEM.02413-06
PubMed Abstract | CrossRef Full Text | Google Scholar
Langenheder, S., and Székely, A. J. (2011). La clasificación de especies y los procesos neutros son importantes durante el montaje inicial de las comunidades bacterianas. ISME J. 5, 1086-1094. doi: 10.1038/ismej.2010.207
PubMed Abstract | CrossRef Full Text | Google Scholar
Liebner, S., Ganzert, L., Kiss, A., Yang, S., Wagner, D., and Svenning, M. M. (2015). Cambios en la composición de la comunidad metanogénica y los flujos de metano a lo largo de la degradación del permafrost discontinuo. Front. Microbiol. 6:356. doi: 10.3389/fmicb.2015.00356
PubMed Abstract | CrossRef Full Text | Google Scholar
Liu, Y. Q., Priscu, J. C., Xiong, J. B., Conrad, R., Vick-Majors, T., Chu, H. Y., et al. (2016). Salinity drives archaeal distribution patterns in high altitude lake sediments on the Tibetan Plateau. FEMS Microbiol. Ecol. 92:fiw033. doi: 10.1093/femsec/fiw033
PubMed Abstract | CrossRef Full Text | Google Scholar
Logares, R., Lindstrom, E. S., Langenheder, S., Logue, J. B., Paterson, H., Laybourn-Parry, J., et al. (2013). Biogeografía de las comunidades bacterianas expuestas a un cambio ambiental progresivo a largo plazo. ISME J. 7, 937-948. doi: 10.1038/ismej.2012.168
PubMed Abstract | CrossRef Full Text | Google Scholar
Loreau, M., Naeem, S., Inchausti, P., Bengtsson, J., Grime, J. P., Hector, A., et al. (2001). Biodiversity and ecosystem functioning: current knowledge and future challenges. Science 294, 804-808. doi: 10.1126/science.1064088
PubMed Abstract | CrossRef Full Text | Google Scholar
Lozupone, C. A., y Knight, R. (2007). Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U.S.A. 104, 11436-11440. doi: 10.1073/pnas.0611525104
PubMed Abstract | CrossRef Full Text | Google Scholar
Luton, P. E., Wayne, J. M., Sharp, R. J., y Riley, P. W. (2002). El gen mcrA como alternativa al 16S rRNA en el análisis filogenético de poblaciones de metanógenos en vertederos. Microbiology 148, 3521-3530. doi: 10.1099/00221287-148-11-3521
PubMed Abstract | CrossRef Full Text | Google Scholar
Lutz, S., Anesio, A. M., Raiswell, R., Edwards, A., Newton, R. J., Gill, F., et al. (2016). La biogeografía de los microbiomas de la nieve roja y su papel en el derretimiento de los glaciares árticos. Nat. Commun. 7:11968. doi: 10.1038/ncomms11968
PubMed Abstract | CrossRef Full Text | Google Scholar
Martiny, J. B. H., Bohannan, B. J. M., Brown, J. H., Colwell, R. K., Fuhrman, J. A., Green, J. L., et al. (2006). Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4, 102-112. doi: 10.1038/Nrmicro1341
PubMed Abstract | CrossRef Full Text | Google Scholar
Martiny, J. B. H., Eisen, J. A., Penn, K., Allison, S. D., y Horner-Devine, M. C. (2011). Drivers of bacterial beta-diversity depend on spatial scale. Proc. Natl. Acad. Sci. U.S.A. 108, 7850-7854. doi: 10.1073/pnas.1016308108
PubMed Abstract | CrossRef Full Text | Google Scholar
McArdle, B. H., y Anderson, M. J. (2001). Fitting multivariate models to community data: a comment on distance-based redundancy analysis. Ecology 82, 290-297. doi: 10.1890/0012-9658(2001)0822.0.CO;2
CrossRef Full Text | Google Scholar
McCalley, C. K., Woodcroft, B. J., Hodgkins, S. B., Wehr, R. A., Kim, E.-H., Mondav, R., et al. (2014). Dinámica del metano regulada por la respuesta de la comunidad microbiana al deshielo del permafrost. Nature 514, 478-481. doi: 10.1038/nature13798
PubMed Abstract | CrossRef Full Text | Google Scholar
McGenity, T. J. (2010). «Methanogens and Methanogenesis in Hypersaline Environments», en Handbook of Hydrocarbon and Lipid Microbiology, ed. K. N. Timmis (Berlín: Springer), 665-680. doi: 10.1007/978-3-540-77587-4_53
CrossRef Full Text | Google Scholar
McLusky, D. S., y Elliott, M. (2004). The Estuarine Ecosystem: Ecology, Threats and Management. New York, NY: Oxford University Press. doi: 10.1093/acprof:oso/9780198525080.001.0001
CrossRef Full Text | Google Scholar
Megonigal, J. P., Mines, M. E., y Visscher, P. T. (2005). «Anaerobic metabolism: linkages to trace gases and aerobic processes», en Biogeochemistry, ed. W. H. Schlesinger (Oxford: Elsevier), 350-362.
Google Scholar
Milferstedt, K., Youngblut, N. D., y Whitaker, R. J. (2010). Spatial structure and persistence of methanogen populations in humic bog lakes. ISME J. 4, 764-776. doi: 10.1038/Ismej.2010.7
PubMed Abstract | CrossRef Full Text | Google Scholar
Mosier, A. C., y Francis, C. A. (2008). Abundancia relativa y diversidad de archaea y bacterias oxidantes de amoníaco en el estuario de la Bahía de San Francisco. Environ. Microbiol. 10, 3002-3016. doi: 10.1111/j.1462-2920.2008.01764.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Nazaries, L., Murrell, J. C., Millard, P., Baggs, L., and Singh, B. K. (2013). Metano, microbios y modelos: comprensión fundamental del ciclo del metano del suelo para futuras predicciones. Environ. Microbiol. 15, 2395-2417. doi: 10.1111/1462-2920.12149
PubMed Abstract | CrossRef Full Text | Google Scholar
Nelson, M. B., Martiny, A. C., y Martiny, J. B. H. (2016). Biogeografía global de los rasgos microbianos de reciclaje de nitrógeno en el suelo. Proc. Natl. Acad. Sci. U.S.A. 113, 8033-8040. doi: 10.1073/pnas.1601070113
PubMed Abstract | CrossRef Full Text | Google Scholar
Nemergut, D. R., Costello, E. K., Hamady, M., Lozupone, C., Jiang, L., Schmidt, S. K., et al. (2011). Global patterns in the biogeography of bacterial taxa. Environ. Microbiol. 13, 135-144. doi: 10.1111/j.1462-2920.2010.02315.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Nobu, M. K., Narihiro, T., Kuroda, K., Mei, R., and Liu, W. T. (2016). Chasing the elusive Euryarchaeota class WSA2: genomes reveal a un metanógeno reductor de metilo exclusivamente fastidioso. ISME J. 10, 2478-2487. doi: 10.1038/ismej.2016.33
PubMed Abstract | CrossRef Full Text | Google Scholar
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O’Hara, R. B., et al. (2015). vegan: Community Ecology Package. R Package Version 2.2.0. Disponible en: https://CRAN.R-project.org/package=vegan
Google Scholar
Oloo, F., Valverde, A., Quiroga, M. V., Vikram, S., Cowan, D., y Mataloni, G. (2016). La heterogeneidad del hábitat y la conectividad dan forma a las comunidades microbianas en las turberas de América del Sur. Sci. Rep. 6:25712. doi: 10.1038/srep25712
PubMed Abstract | CrossRef Full Text | Google Scholar
Pattnaik, P., Mishra, S. R., Bharati, K., Mohanty, S. R., Sethunathan, N., and Adhya, T. K. (2000). Influence of salinity on methanogenesis and associated microflora in tropical rice soils. Microbiol. Res. 155, 215-220. doi: 10.1016/S0944-5013(00)80035-X
PubMed Abstract | CrossRef Full Text | Google Scholar
Poffenbarger, H. J., Needelman, B. A., y Megonigal, J. P. (2011). Influencia de la salinidad en las emisiones de metano de las marismas. Wetlands 31, 831-842. doi: 10.1007/s13157-011-0197-0
PubMed Abstract | CrossRef Full Text | Google Scholar
PubMed Abstract | CrossRef Full Text | Google Scholar
R Core Team (2014). R: Un lenguaje y entorno para la computación estadística (versión 3.0.3). Viena: R Foundation for Statistical Computing.
Google Scholar
Roberts, D. (2016). labdsv: Ordination and Multivariate Analysis for Ecology. R Package Version 1.8-0. Disponible en: https://CRAN.R-project.org/package=labdsv
Google Scholar
Rooney-Varga, J. N., Giewat, M. W., Duddleston, K. N., Chanton, J. P., y Hines, M. E. (2007). Links between archaeal community structure, vegetation type and methanogenic pathway in Alaskan peatlands. FEMS Microbiol. Ecol. 60, 240-251. doi: 10.1111/j.1574-6941.2007.00278.x
PubMed Abstract | CrossRef Full Text | Google Scholar
Rosenberg, E., DeLong, E. F., Lory, S., Stackebrandt, E., y Thompson, F. (2014). The Prokaryotes: Otros Linajes Mayores de Bacterias y las Arqueas. Berlín: Springer.
Google Scholar
Ruff, S. E., Biddle, J. F., Teske, A. P., Knittel, K., Boetius, A., y Ramette, A. (2015). Dispersión global y diversificación local del microbioma de las filtraciones de metano. Proc. Natl. Acad. Sci. U.S.A. 112, 4015-4020. doi: 10.1073/pnas.1421865112
PubMed Abstract | CrossRef Full Text | Google Scholar
Schauer, R., Bienhold, C., Ramette, A., y Harder, J. (2009). Bacterial diversity and biogeography in deep-sea surface sediments of the South Atlantic Ocean. ISME J. 4, 159-170. doi: 10.1038/ismej.2009.106
PubMed Abstract | CrossRef Full Text | Google Scholar
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537-7541. doi: 10.1128/Aem.01541-09
PubMed Abstract | CrossRef Full Text | Google Scholar
Statham, P. J. (2012). Nutrientes en los estuarios – una visión general y los impactos potenciales del cambio climático. Sci. Total Environ. 434, 213-227. doi: 10.1016/j.scitotenv.2011.09.088
PubMed Abstract | CrossRef Full Text | Google Scholar
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579-591. doi: 10.1038/Nrmicro1931
PubMed Abstract | CrossRef Full Text | Google Scholar
Tilman, D., Isbell, F., and Cowles, J. M. (2014). Biodiversidad y funcionamiento de los ecosistemas. Annu. Rev. Ecol. Evol. Syst. 45, 471-493. doi: 10.1146/annurev-ecolsys-120213-091917
CrossRef Full Text | Google Scholar
Ulrich, W., Hajdamowicz, I., Zalewski, M., Stañska, M., Ciurzycki, W., and Tykarski, P. (2010). Species assortment or habitat filtering: a case study of spider communities on lake islands. Ecol. Res. 25, 375-381. doi: 10.1007/s11284-009-0661-y
CrossRef Full Text | Google Scholar
Vanwonterghem, I., Evans, P. N., Parks, D. H., Jensen, P. D., Woodcroft, B. J., Hugenholtz, P., et al. (2016). Methylotrophic methanogenesis discovered in the archaeal phylum Verstraetearchaeota. Nat. Microbiol. 1:16170. doi: 10.1038/nmicrobiol.2016.170
PubMed Abstract | CrossRef Full Text | Google Scholar
von Mering, C., Hugenholtz, P., Raes, J., Tringe, S. G., Doerks, T., Jensen, L. J., et al. (2007). Quantitative phylogenetic assessment of microbial communities in diverse environments. Science 315, 1126-1130. doi: 10.1126/science.1133420
PubMed Abstract | CrossRef Full Text | Google Scholar
Wagner, D., and Liebner, S. (2009). «Global warming and carbon dynamics in permafrost soils: methane production and oxidation», en Permafrost Soils, ed. R. Margesin (Berlín: Springer), 219-236.
Google Scholar
Weiher, E., y Keddy, P. (2001). Reglas de la asamblea ecológica: Perspectivas, avances, retrocesos. Cambridge: Cambridge University Press.
Google Scholar
Whitaker, R. J., Grogan, D. W., y Taylor, J. W. (2003). Las barreras geográficas aíslan poblaciones endémicas de arqueas hipertermófilas. Science 301, 976-978. doi: 10.1126/science.1086909
PubMed Abstract | CrossRef Full Text | Google Scholar
Wickham, H. (2009). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer-Verlag. doi: 10.1007/978-0-387-98141-3
CrossRef Full Text | Google Scholar
Yang, J., Ma, L. A., Jiang, H., Wu, G., and Dong, H. (2016). La salinidad moldea la diversidad microbiana y la estructura de la comunidad en los sedimentos superficiales de los lagos Qinghai-Tibetanos. Sci. Rep. 6:25078. doi: 10.1038/srep25078
PubMed Abstract | CrossRef Full Text | Google Scholar
Yang, S., Liebner, S., Alawi, M., Ebenhöh, O., and Wagner, D. (2014). Base de datos taxonómica y valor de corte para el procesamiento de datos de pirosecuenciación del gen mcrA 454 por MOTHUR. J. Microbiol. Methods 103, 3-5. doi: 10.1016/j.mimet.2014.05.006
PubMed Abstract | CrossRef Full Text | Google Scholar
Yang, S., Liebner, S., Winkel, M., Alawi, M., Horn, F., Dörfer, C., et al. (2017). Análisis en profundidad de las comunidades metanogénicas del núcleo de los humedales afectados por el permafrost a gran altura. Soil Biol. Biochem. 111, 66-77. doi: 10.1016/j.soilbio.2017.03.007
CrossRef Full Text | Google Scholar
Ye, R. Z., Jin, Q. S., Bohannan, B., Keller, J. K., McAllister, S. A., and Bridgham, S. D. (2012). pH controls over anaerobic carbon mineralization, the efficiency of methane production, and methanogenic pathways in peatlands across an ombrotrophic-minerotrophic gradient. Soil Biol. Biochem. 54, 36-47. doi: 10.1016/j.soilbio.2012.05.015
CrossRef Full Text | Google Scholar
Zinder, S. H. (1993). «La ecología fisiológica de methanogens,» en Methanogenesis: Ecology, Physiology, Biochemistry & Genetics, ed. J. G. Ferry (Boston, MA: Springer), 128-206.
Google Scholar
Zinger, L., Amaral-Zettler, L. A., Fuhrman, J. A., Horner-Devine, M. C., Huse, S. M., Welch, D. B. M., et al. (2011). Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE 6:e24570. doi: 10.1371/journal.pone.0024570
PubMed Abstract | CrossRef Full Text | Google Scholar