Neste post introduzimos os carbamatos como um útil grupo protetor para aminas, particularmente no contexto de fazer peptídeos.
Aqui está um resumo rápido do que iremos abordar neste post, com mais detalhes abaixo.
Table of Contents
- An Introduction To Simple Peptide Synthesis
- Let’s Mix Up A Batch of Gly-Ala
- A Very Bad Initial Plan
- “Hark! Que porcaria é esta?”
- Um chapéu protector para NH2
- Mate! Use Um Carbamato, Mate!
- Boc e CBz Are The Bees Knees
- Instalação e Remoção do Grupo Protector “Boc”
- Instalação e Remoção do Grupo Protector de CBz (ou “Z”) Carbamato
- Uma Síntese Simples de Peptídeo Usando Grupos Protectores de Carbamato
- Mantenha-se…
- Notas
- (Avançado) Referências e Leituras Adicionais
>
>
>
>
Uma Introdução à Síntese Simples do Peptídeo
Num post recente passamos por 3 formas comuns de fazer amides:
- Adicionamento de aminas a halogenetos de acilo / anidridos
- Hidrólise parcial de nitrilos
- Acoplamento de ácidos carboxílicos com aminas usando um agente desidratante como DCC (N,N’-diciclohexylcarbodiimida).
O que faltava naquele post era qualquer menção de sintetizar o bisavô das ligações amídicas mais úteis conhecidas pela humanidade, e com isso quero dizer os peptídeos.
Uma ligação peptídeo é o nome que damos à ligação amida que une dois aminoácidos.
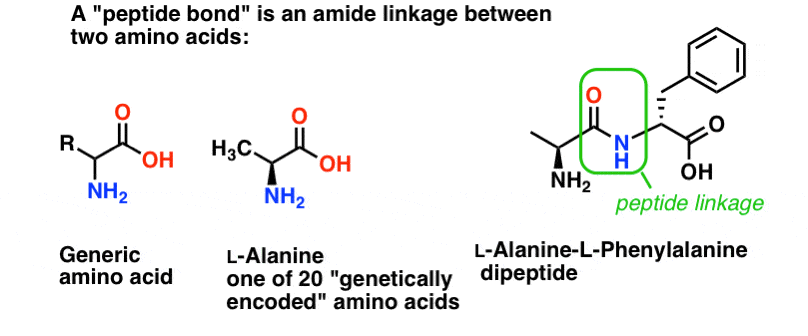
E os aminoácidos são importantes porque… espere. Você já sabe porque os aminoácidos são importantes, certo? Se não sabe isto agora, volte depois de ler isto.
Vamos misturar um lote de Gly-Ala
Tentemos aplicar algumas das nossas novas habilidades de síntese de amida para tentar construir um dipeptídio muito simples, a glicina-alanina. Se pudermos construir um dipeptídio simples agora, podemos usar o que aprendemos para mostrar como peptídeos ainda mais complexos são feitos em um post posterior. Você sabia que o vencedor do Prêmio Nobel Bruce Merrifield sintetizou a insulina unindo um aminoácido de cada vez? Sim, realmente.
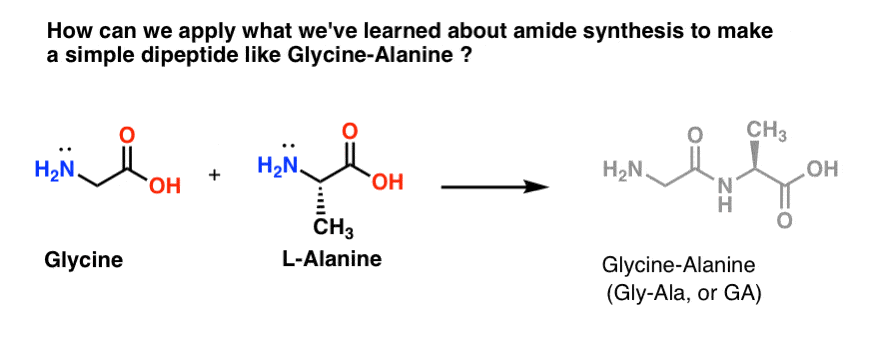
Dos três métodos que listamos para fazer amidas, apenas dois são potencialmente úteis para formar ligações de peptídeos: 1) o método do cloreto ácido, ou 2) síntese através de um agente de acoplamento como o DCC. .
3. um plano inicial muito ruim
Nossa primeira facada na síntese do peptídeo envolverá barrelling para frente com um plano de ataque muito ingênuo e esperar que tudo funcione no final. Segurem-se, pois vai ficar confuso.
Vamos começar propondo o método do cloreto ácido para a síntese de Gly-Ala.
Olhando para trás a partir do dipeptídio de Gly-Ala, nosso plano nos faria sintetizar a ligação do peptídeo através da reação da alanina com o cloreto ácido derivado da glicina:
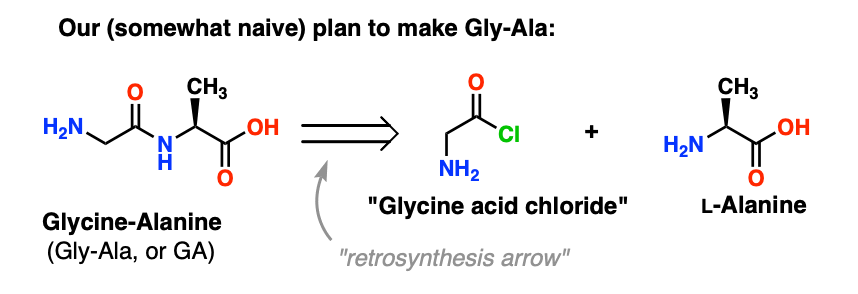
Há um pequeno problema com este plano. Pode ser um pouco difícil de detectar no início.
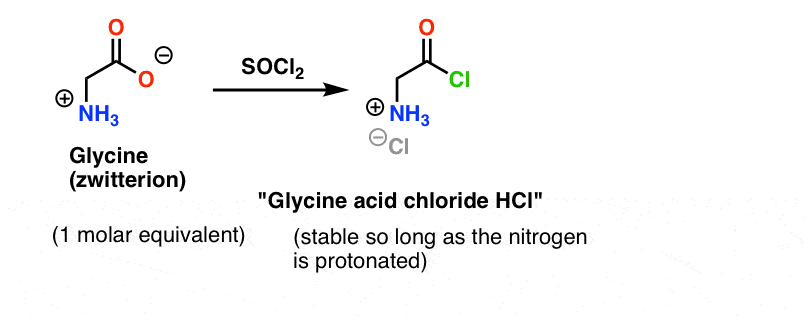
Vamos assumir que fizemos o cloreto ácido de glicina* , e temos “cloreto ácido de glicina” e alanina juntos no mesmo frasco, juntamente com algum excesso de base para acelerar as coisas ao longo.
Realizaremos a reacção na direcção da frente:
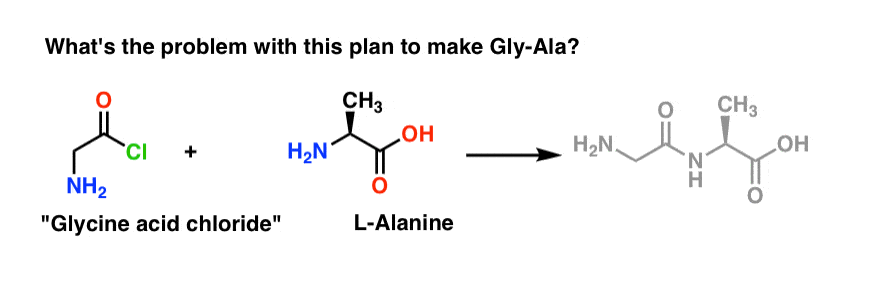
O que poderia correr mal?
“Ouçam! Que porcaria é esta? “
O nosso plano é fazer uma solução de cloreto ácido de glicina (equivalente a 1 molar), e depois tê-la pacientemente à espera no frasco até adicionarmos um equivalente molar de alanina, e então ele reagiria com o grupo nucleofílico NH2 de alanina.
O problema com nosso plano é que não estamos lidando com uma única molécula de “cloreto ácido glicina” – estamos lidando com algo em torno de uma molécula (6,02 x 1023 moléculas) dela. E o cloreto ácido de glicina já tem um grupo nucleofílico NH2!
“Cloreto ácido de glicina” como desenhado, não é uma molécula estável, porque pode reagir com ela mesma.
Isto significa que uma solução de cloreto ácido de glicina deixada à sua própria disposição formaria um polímero de glicina, com a estrutura Glica-Lica-Lica-Lica-…
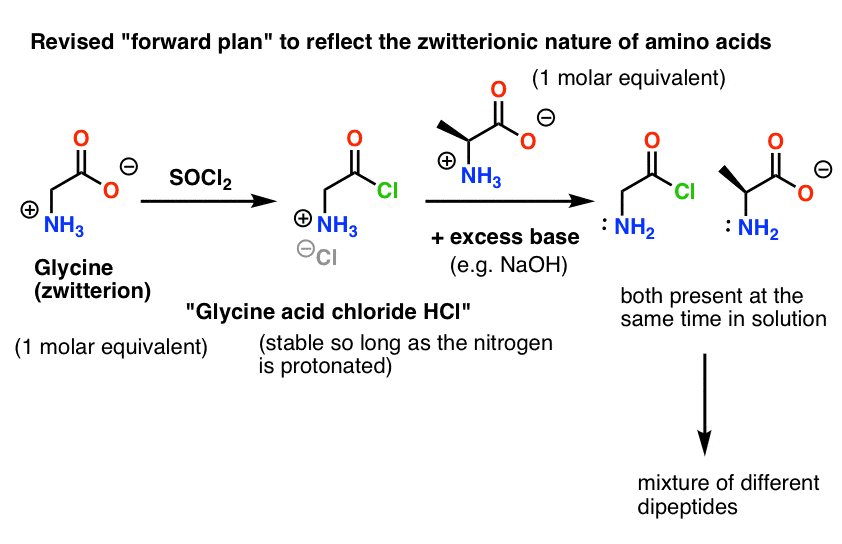
Even uma solução de cloreto ácido de glicina na presença de alanina formaria não só o desejado Gly-Ala, mas também Gly-Gly (com um halogeneto de acilo preso) que pode continuar a fazer mais maldades com outro nucleófilo, seja Gly ou Ala:
(e não, não há muito para distinguir o NH2 de “cloreto de ácido glicínico” do NH2 de alanina. Eles são igualmente reactivos.)
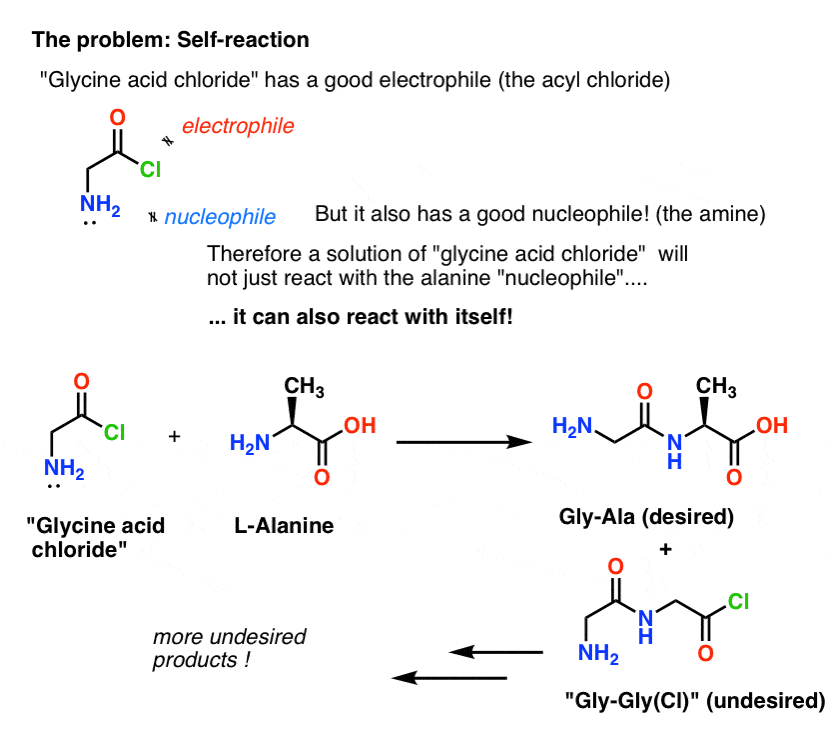
A lição aqui é que quando se tem uma solução de uma molécula contendo tanto um nucleófilo como um electrófilo, ela pode auto-reagir. Há um nome para este processo que pode soar familiar: polimerização.
Então, como impedimos que isto aconteça?
Um chapéu protector para NH2
A melhor maneira é “tapar” o nitrogénio de alguma forma com um grupo protector (PG) que torna o grupo NH2 não-nucleofílico. Ele também deve ter as seguintes propriedades:
- ser fácil e seletivamente instalado
- interferir as condições de reação desejadas (ex. SOCl2 para fazer o cloreto ácido a partir do ácido carboxílico)
- suavemente e selectivamente removido sem afectar o produto final
Já vimos estratégias de protecção de grupo, particularmente com álcoois e o uso de reagentes Grignard.
Aqui está como poderia ser uma estratégia de protecção de grupo para a nossa síntese de “Gly-Gly”. Instalamos um grupo protetor (“PG”) na glicina, depois fazemos o cloreto ácido. O PG deve ser escolhido de forma a tornar o nitrogênio não-nucleofílico (ou seja, ele não reagirá com o cloreto ácido).
Podemos então formar nossa ligação peptídeo com a alanina desprotegida e então remover o PG sob condições suaves.
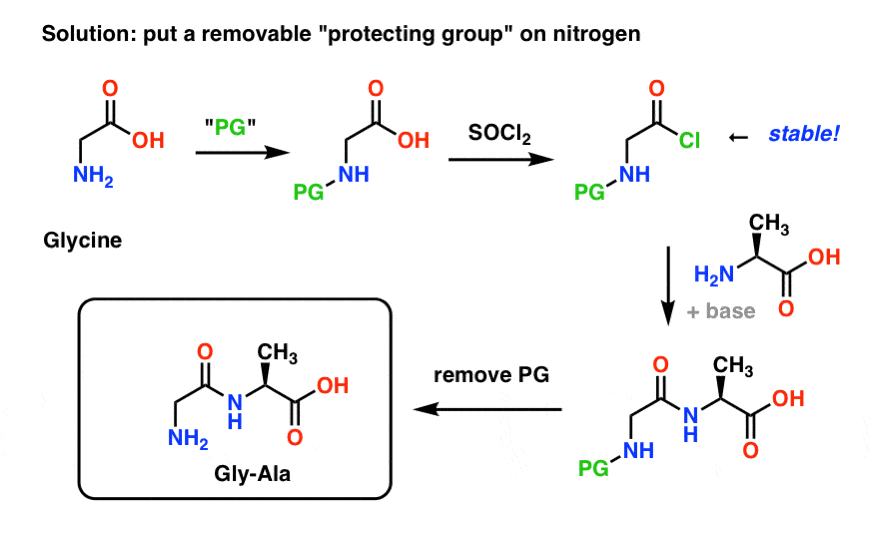
Uma estratégia de grupo protetor para nitrogênio que já exploramos é a síntese de Gabriel, que usa ftalimida (pode-se pensar em um ftalimida como nitrogênio protegido). Isto tem sido utilizado para sintetizar a Glicoglidina (“glycylglycine”) ! Um dos problemas, entretanto, é que condições relativamente severas (calor amplo) são necessárias para instalar e remover o grupo ftalimida, e este não é um ambiente muito saudável para a sobrevivência de aminoácidos quirais sensíveis, que podem facilmente racemizar.
Outra escolha potencial é proteger o nitrogênio como amida, mas amidas de clivagem também podem requerer condições severas. Além disso, como estamos tentando forjar uma ligação amida (peptídeo) aqui de qualquer forma, podemos ter problemas de seletividade com sua remoção – destruindo a vila para salvá-la.
Mate! Use Um Carbamato, Mate!
A escolha mais popular do grupo de proteção do nitrogênio amina é o grupo funcional do carbamato. Um carbamato parece a criança bastarda de um éster e uma amida, com N e O flanqueando um carbonilo.
O nitrogênio de um carbamato é relativamente não-nucleofílico, e além disso, os carbamatos são:
- facilmente instalado sobre azoto
- inerte a uma grande variedade de condições de reacção
- facilmente removido sem afectar os grupos amida existentes
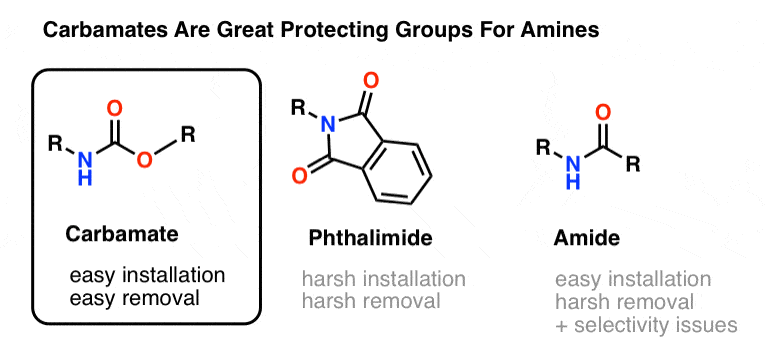
Isto torna-os perfeitos para os nossos propósitos.
Boc e CBz Are The Bees Knees
Dois populares grupos protetores de carbamato são Boc (t-Butyloxycarbonyl) e CBz (carboxybenzyl).
Para os nossos propósitos, estes dois grupos protetores podem ser considerados como mais ou menos equivalentes, pois ambos podem ser usados efetivamente para síntese de peptídeos.
A diferença chave está realmente em como eles são removidos (ou seja, o passo da “desproteção”). A escolha entre uma ou outra torna-se crucial quando se tem uma molécula complexa com múltiplos grupos de protecção; isso enquadra-se na categoria de “estratégia sintética avançada”, que é mais um assunto para o Org 3.
Instalação e Remoção do Grupo Protector “Boc”
O grupo Boc é normalmente instalado com “Boc2O” (por vezes referido como “anidrido Boc”), e é removido com ácido. A escolha habitual é o ácido trifluoroacético (TFA) “puro” (ou seja, não diluído), que liberta os grupos Boc de forma muito limpa, libertando CO2 e álcool t-butílico.
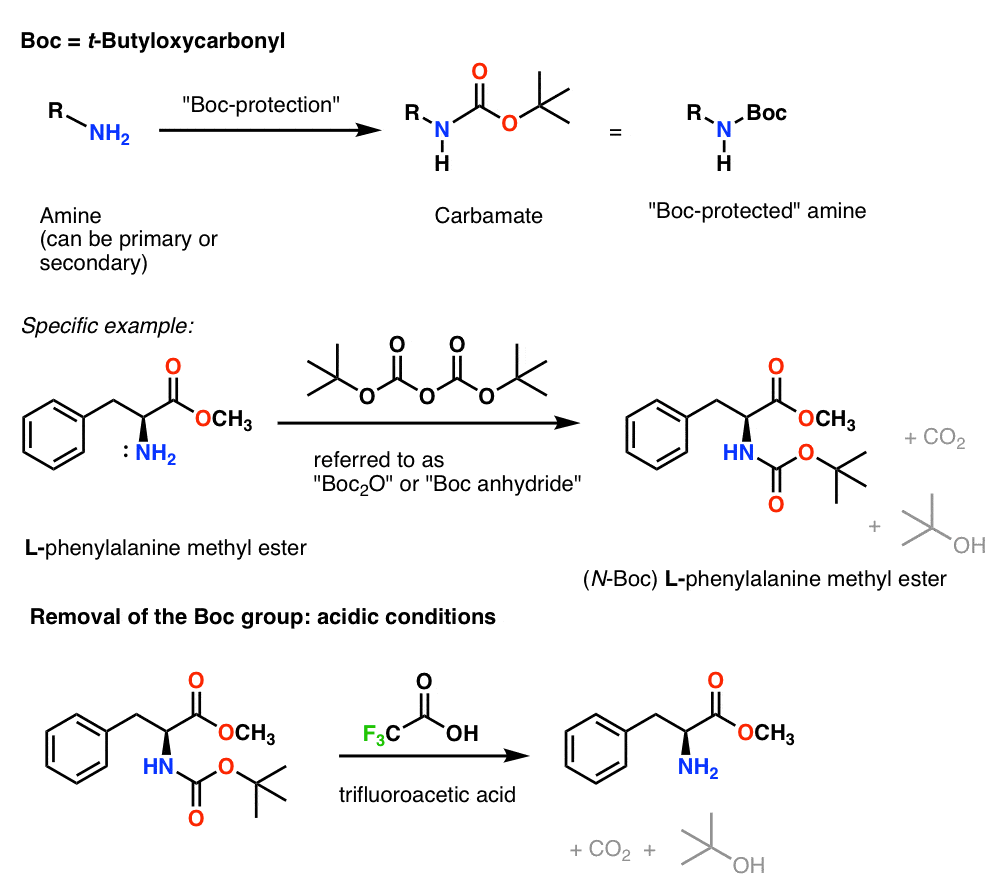
Instalação e remoção do grupo protetor de carbamato de CBz (ou “Z”)
O grupo Cbz (às vezes abreviado como “Z”) pode ser instalado com CbzCl e base leve, e normalmente é removido por hidrogenação catalítica (Pd-C/H2). Isto é extremamente suave e tem a vantagem de ocorrer em pH neutro, deixando os grupos funcionais sensíveis a ácido ou base sozinhos.
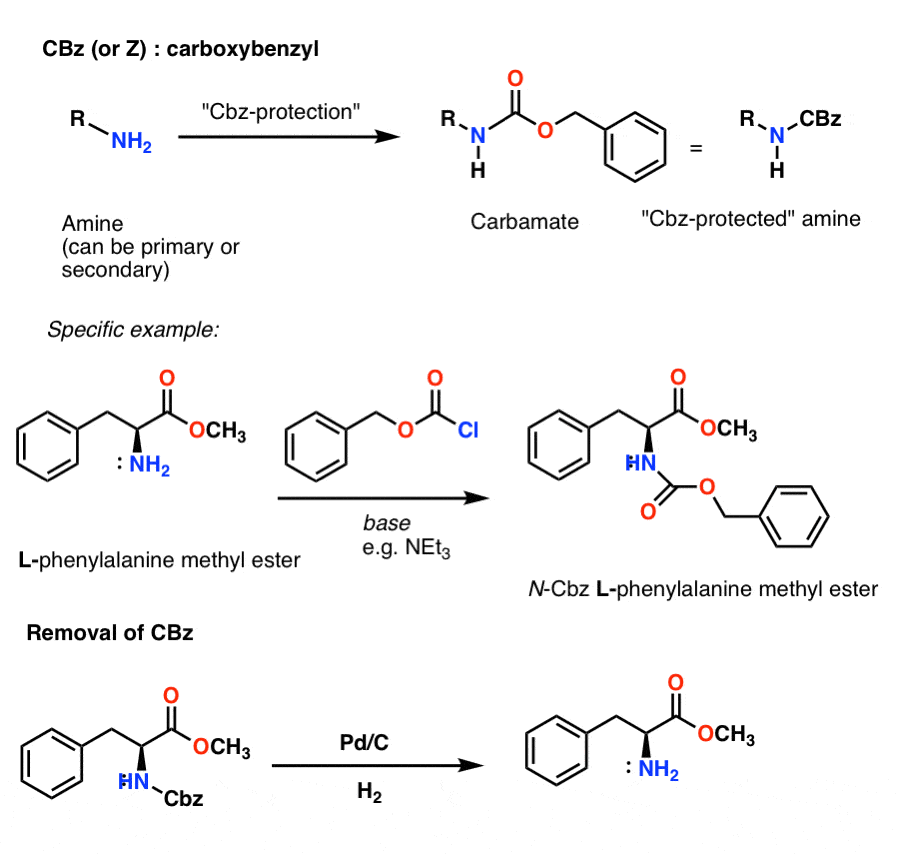
A Simple Peptide Synthesis Using Carbamate Protecting Groups
Vamos voltar à síntese do peptídeo e aplicar esta estratégia de grupo protetor para fazer Gly-Ala.
Comecemos com um aminoácido como a L-alanina. Tratando a alanina com Boc2O, obtemos a L-alanina protegida com N-Boc. O passo seguinte é formar um cloreto ácido usando SOCl2. Uma vez formada, adicionamos nossa amina (por exemplo, L-valina) na presença de excesso de base, formando nossa ligação amida chave. O passo final para dar o dipeptídeo é desproteger a amina Boc protegida com ácido trifluoroacético (TFA), e voilá! temos o nosso dipeptídeo.
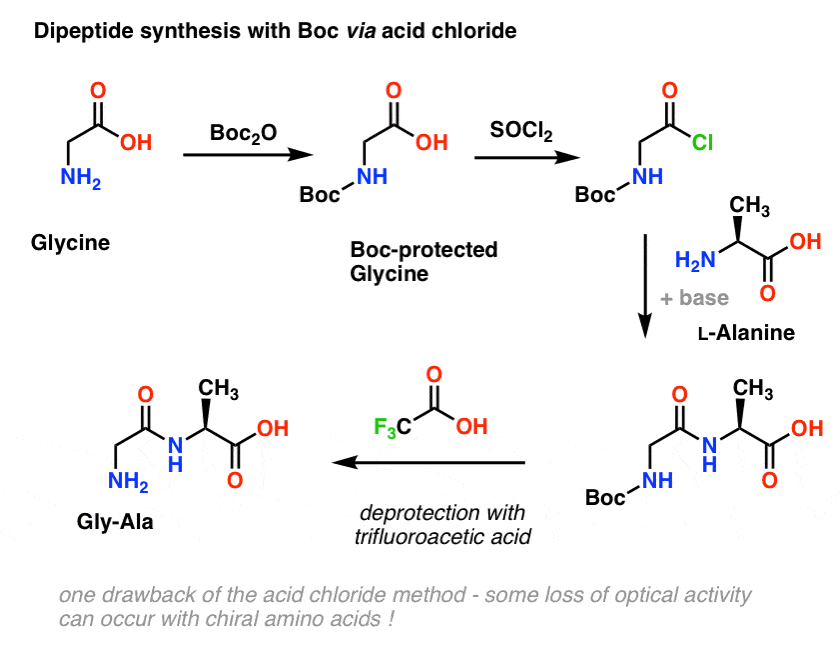
Embora este método possa ser bom no papel, um problema do uso de cloretos ácidos na prática é que os aminoácidos quirais frequentemente perdem sua pureza óptica através deste método, um processo às vezes referido como “racemização”, mas mais corretamente chamada “epimerização” (tecnicamente mais correta, porque um hidrogênio em um centro quiral está invertido)
Desde que a quiralidade dos aminoácidos é essencial para sua função biológica, um protocolo ligeiramente mais suave é geralmente usado que emprega DCC ou um reagente de acoplamento similar.
Aqui, tratamos a glicina Boc-protegida com DCC para activar o ácido carboxílico. Depois adicionamos o nosso aminoácido nucleófilo (L-alanina) que forma o dipeptídeo. Se quisermos isolar o dipeptídeo Gly-Ala neste ponto, podemos então remover o grupo Boc com TFA.
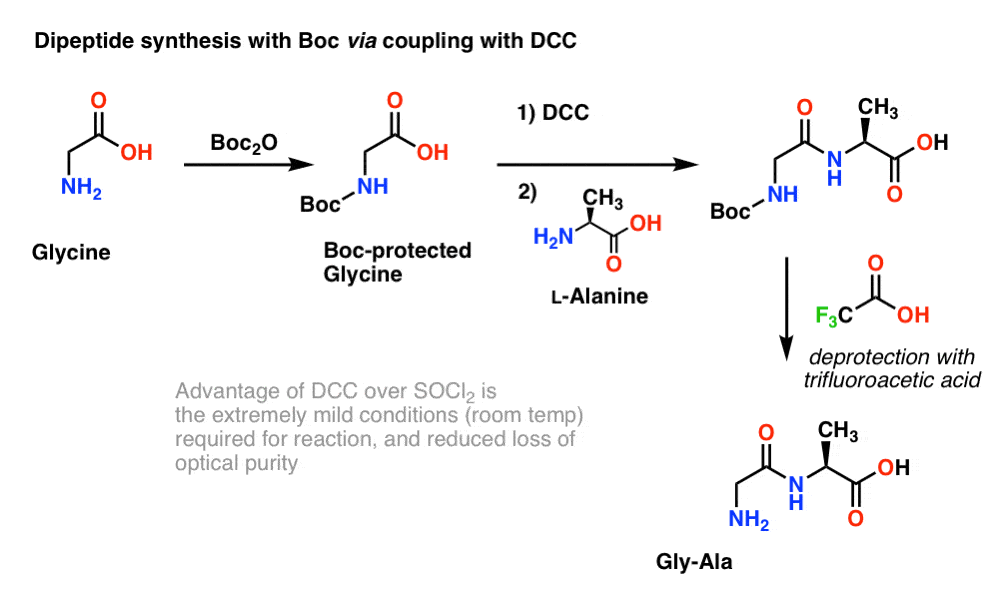
(nota sobre este esquema)
Mantenha-se…
Notem que se quisermos fazer um tri-peptídeo, podemos continuar a realizar ciclos de adição de DCC (para activar o ácido carboxílico) seguidos da adição de novos aminoácidos, construindo o peptídeo uma unidade de cada vez!
Existe um método particularmente eficaz para construir peptídeos mais longos, pioneiro por Bruce Merrifield (e aplicado na síntese de insulina, entre outros) chamado de síntese de peptídeo de fase sólida, que cobriremos na próxima vez que estivermos sobre este tópico.
Notas
Nota 1. Similar à velha piada sobre o economista que propõe um plano para sair de uma ilha deserta:
“Assuma que temos um barco”.
Glycine (como todos os aminoácidos) em si é um zwitterion. O tratamento da glicina com SOCl2 deve produzir o cloreto ácido com uma amina protonada. Esta deve ser relativamente estável em solução desde que não seja adicionada nenhuma base.
Aqui está o problema. Como nosso aminoácido nucleófilo (alanina) também é zwitterionico, nenhuma reação pode ocorrer até que o excesso de base seja adicionado para liberar um par solitário sobre o nitrogênio alanino. Após a adição da base, temos “cloreto ácido glicina” e alanina juntos em solução. Não há diferença significativa na nucleofilia entre os nitrogênios destas duas espécies, e cada um deles irá competir para reagir com o nucleófilo ácido clorídrico, levando a uma mistura de Gly-Ala e Gly-GlyCl, e o Gly-GlyCl pode então reagir ainda mais com os vários nucleófilos predefinidos em solução para dar peptídeos tri-, tetra- e superiores.
Nota 2. John Sheehan, que conhecemos anteriormente como o inventor do DCC a caminho do primeiro a sintetizar a penicilina, também fez Gly-Gly protegido contra ftalil através de uma síntese de Gabriel:
Referência aqui (JACS, 1949, 71, 1856)
Nota 3. Outro problema com o uso de grupos protetores de amida é a formação de azlactona, que pode levar à epimerização de aminoácidos quirais. Veja também este conjunto de problemas.
Nota 4. “Grupos Ortogonais Protectores”. No planejamento sintético é crucial ter grupos protetores que são removíveis sob condições distintamente diferentes. Esta propriedade é frequentemente referida como “ortogonalidade”
Por exemplo, no dipeptídio seguinte temos dois grupos de protecção diferentes sobre azoto – um Boc e um CBz. Selecionando grupos protetores “ortogonais”, cada nitrogênio é endereçável – podemos escolher qual grupo protetor remover, e nossa síntese pode prosseguir a partir daí. Isto evita entrar numa situação em que temos duas aminas desprotegidas e temos de confiar que uma é mais reactiva do que a outra. Estas abordagens muito raramente funcionam!
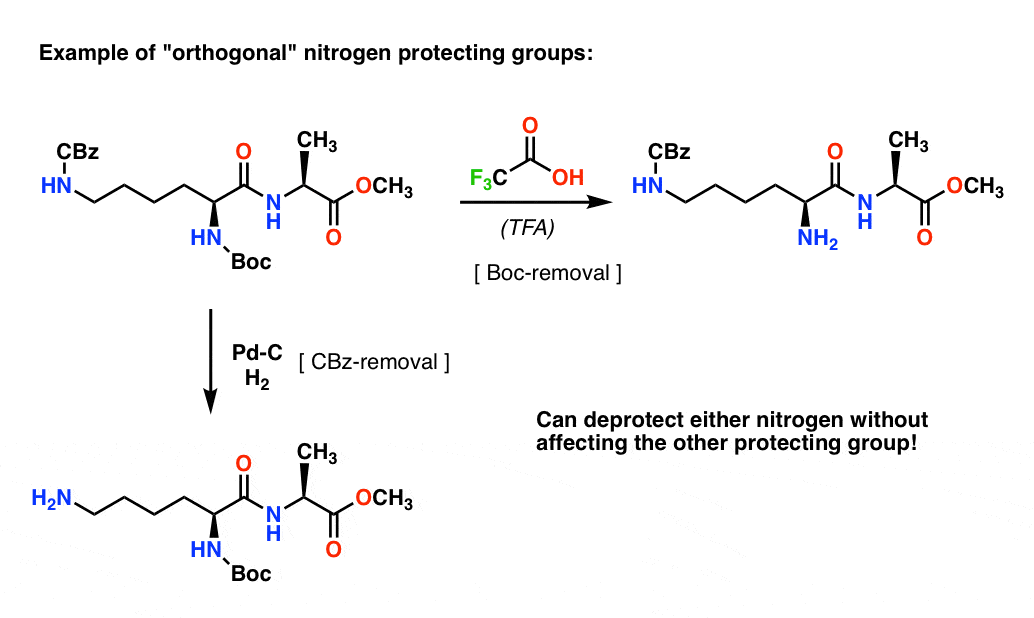
Uma nota – para simplificar aqui a alanina é retratada com um ácido carboxílico livre, mas uma abordagem ligeiramente melhor seria usar o éster metílico de alanina para evitar qualquer auto-acoplamento entre a amina livre de alanina e o ácido carboxílico livre.
Mate…
Referências (Avançadas) e Leitura Adicional
Carbamatos são úteis como grupos protetores para aminas, e os mais comumente empregados são -Boc, -Cbz, e -Fmoc.
- Über ein allgemeines Verfahren der Peptid-Synthese
Max Bergmann e Leonidas Zervas
Ber. 1932, 65 (7), 1192-1201
DOI: 10.1002/cber.19320650722
O grupo de proteção -Cbz (carboxybenzyl) foi usado pela primeira vez por Max Bergmann e Leonidas Zervas em 1932 para a síntese de peptídeos, e às vezes é abreviado “-Z” em homenagem a Zervas. - REMOÇÃO DE GRUPOS PROTECTORES DE T-BUTYL E T-BUTOXICARBONILO COM ÁCIDO TRIFLUOROACETICO
Mecanismos, Formação de Biprodutos e Avaliação de Catadores
Behrend F. Lundt, Nils L. Johansen, Aage Vølund, e Jan Markussen
J. Pept. Prot. Res. 1978, 12 (5), 258-268
DOI: 10.1111/j.1399-3011.1978.tb02896.x
Na prática, os necrófagos nucleófilos (por exemplo, thiols) são normalmente adicionados ao coquetel ácido (TFA) quando desprotegidos, uma vez que a desproteção de Boc dará espécies eletrofílicas de t-butil (por exemplo, t-butil trifluoroacetato) que podem reagir com resíduos sensíveis (por exemplo, Trp ou Cys). - Método de Síntese de Cadeias Longas de Peptídeo Usando uma Síntese de Oxitocina como Exemplo
Miklos Bodanszky e Vincent du Vigneaud
Journal of the American Chemical Society 1959, 81 (21), 5688-5691
DOI: 1021/ja01530a040
Na primeira metade do século 20, a síntese do peptídeo foi feita usando técnicas de fase de solução química orgânica padrão. Esta é agora conhecida como LPPS (síntese de peptídeo em fase líquida). du Vigneaud recebeu o Prêmio Nobel de Química em 1955 por seu trabalho em mostrar que a síntese do peptídeo poderia ser alcançada, usando a escolha correta de grupos protetores e estratégias sintéticas. - A FORMAÇÃO TEMPORÁRIA DO ANEL AZLACTONO NA RACEMIZAÇÃO DOS DERIVADOS DE ACIDEZES DE AMINO ACIDENTE COM ANHIDRIDE ÁCTICO
Vincent du Vigneaud e Curtis E. Meyer
Biol. Quimioterapia. 1932, 99:143-151
http://www.jbc.org/content/99/1/143.citation
Outro problema com o uso de grupos protetores de amidas é a formação de azlactona, que pode levar à epimerização de aminoácidos quirais. - Uma Nova Rota Sintética para Peptídeos
John C. Sheehan e Victor S. Frank
Journal of the American Chemical Society 1949, 71 (5), 1856-1861
DOI: 10.1021/ja01173a095
John Sheehan, o inventor do DCC a caminho da primeira síntese de penicilina, também fez Gly-Gly protegido contra ftalil através de uma síntese de Gabriel. - Um Novo Método de Formação de Títulos de Peptídeo
John C. Sheehan e George P. Hess
Journal of the American Chemical Society 1955, 77 (4), 1067-1068
DOI: 1021/ja01609a099
Papel original sobre a síntese de ligações peptídeo/amida usando DCC. - Função 9-Fluorenilmetoxicarbonilo, um novo grupo protetor amino-base sensível
Louis A. Carpino e Grace Y. Han
Journal of the American Chemical Society 1970, 92 (19), 5748-5749
DOI: 10.1021/ja00722a043 - 9-Fluorenilmetoxicarbonil amino-protector grupo
Louis A. Carpino e Grace Y. Han
The Journal of Organic Chemistry 1972 37 (22), 3404-3409
DOI: 10.1021/jo00795a005
A descoberta e o desenvolvimento do grupo de protecção -Fmoc para aminas adiciona outra camada ortogonal às estratégias de protecção/deprotecção das aminas. O grupo -Fmoc é labial de base, e na síntese de peptídeos é normalmente removido com 20% de piperidina em DMF. Cbz é removido por hidrogenação, -Boc removido com ácido, e -Fmoc com base.