1 – Definition
ARVD ist eine Erkrankung des Herzmuskels, die mit ventrikulären Arrhythmien und plötzlichem Tod einhergeht. Sie ist durch strukturelle und funktionelle Anomalien des rechten Ventrikels gekennzeichnet, die durch den Ersatz des Myokards durch Fett- und Fasergewebe verursacht werden. Die Stellen, an denen die anatomischen Anomalien auftreten, befinden sich im so genannten Dysplasiedreieck (die rechtsventrikulären subtricuspidalen Bereiche, der Apex und das Infundibulum)(4).
Die arrythmogene rechtsventrikuläre Dysplasie (ARVD) ist eine Erbkrankheit, die typischerweise als autosomal-dominantes Merkmal mit unterschiedlicher Penetranz und unvollständiger Ausprägung vererbt wird(1). Es gibt eine autosomal rezessive Variante, die mit palmoplantarer Keratose und schütterem Haar assoziiert ist und als Naxos-Krankheit bezeichnet wird.
2 – Prävalenz
Die Prävalenz in der Allgemeinbevölkerung liegt bei etwa 1:2500 bis 1:5000
Allerdings ist sie von den geographischen Gegebenheiten abhängig, so ist die Prävalenz in bestimmten Regionen Italiens (Padua, Venedig) und Griechenlands (Insel Naxos) erhöht(2). Sie macht 5 bis 10 % der ungeklärten plötzlichen Herztode bei Personen unter 65 Jahren aus(3).
Sie tritt bei jungen Erwachsenen mit einem Verhältnis von Männern zu Frauen von 2,7/1 auf. Nach der hypertrophen Herzkrankheit ist sie die häufigste Ursache für den plötzlichen Herztod bei jungen Menschen.
3 – Diagnose
Die Diagnose der ARVD ist oft schwierig, da es keinen einzigen Test gibt. Die Diagnose basiert auf dem Vorhandensein von strukturellen, histologischen, elektrokardiographischen und genetischen Faktoren gemäß dem Bericht der Task Force von McKenna et al. (5) aus dem Jahr 1994 (Tabelle 1) und einer Modifikation der Task Force von Homid et al. (6) (Tabelle 2), um die diagnostische Sensitivität von Familienmitgliedern ersten Grades für die Früherkennung der Krankheit zu erhöhen.
A) Klinische Anamnese
Zunächst beginnen wir mit dem einfacheren Instrument, nämlich der klinischen Anamnese.
Die klinische Präsentation variiert von asymptomatischen Formen bis hin zu Herzklopfen, Müdigkeit, Synkope oder sogar Herzstillstand, in der Regel während der Belastung. Diese Symptome sind auf ventrikuläre Ektopien, anhaltende ventrikuläre Tachykardien mit Linksschenkelblockkonfiguration oder rechtsventrikuläres Versagen zurückzuführen(4).
Im natürlichen Verlauf dieser Krankheit lassen sich vier Stadien unterscheiden(7):
a) die frühe oder stille Phase, eine subklinische Phase mit verborgenen strukturellen Anomalien
b) die instabile Phase mit elektrischen Störungen
c) die Phase des rechtsventrikulären Versagens
d) die Endphase mit fortschreitendem biventrikulärem Versagen, das eine dilatative Kardiomyopathie imitiert
B) Diagnostische Tests
1.- Elektrokardiogramm
Es gibt mehrere EKG-Merkmale, die bei der Diagnose der ARVD eine Rolle spielen:
a) T-Wellen-Inversionen in V1 bis V3 (kleines diagnostisches Kriterium, aber eine der häufigsten EKG-Anomalien, die bei 85 % der Patienten auftritt (8)
b) QRS-Dauer = 110 ms in V1 bis V3
c) Epsilon-Welle (elektrische Potentiale nach dem Ende des QRS-Komplexes). Sie ist ein wichtiges diagnostisches Kriterium, das in bis zu 30 % der Fälle von ARVD gefunden wird.
Weitere EKG-Marker für ARVD wurden berichtet: QRS- und QT-Dispersion, parietaler Block, definiert als eine QRS-Dauer in den Ableitungen V1 bis V3, die die QRS-Dauer in Ableitung V6 um > 25 ms übersteigt, ein verlängerter S-Wellen-Aufwärtshub in V1 bis V3 = 55 ms (dies wurde als das häufigste EKG-Merkmal bei 95 % der ARVD beobachtet8
2.- Myokardiale Bildgebung
2.1 Echokardiographie
Sie ist das am häufigsten eingesetzte nicht-invasive bildgebende Verfahren, ist jedoch bei übergewichtigen Patienten und bei Patienten mit Lungenemphysem nicht immer das optimale bildgebende Verfahren.
Die Echokardiographie ist der erste diagnostische Ansatz bei Patienten mit Verdacht auf ARVD. Die wichtigsten Befunde sind (9):
– rechtsventrikuläre Dilatation und Hypokinese
– isolierte Dilatation des rechtsventrikulären Ausflusstraktes
– erhöhte Reflektivität des Moderators
– enddiastolische Aneurysmen
– Akinese-Dyskinese des inferobasalen Segments und des rechtsventrikulären Apex
– prominente apikale Trabekel
2.2 Rechtsventrikuläre Kontrastmittelangiographie
Dieses Verfahren gilt als Referenzstandard für die Diagnose der ARVD (10). Sie besteht aus akinetisch-dyskinetischen Bereichen, die im anatomischen Dreieck der Dysplasie lokalisiert sind. Aufgrund der invasiven Technik, der Röntgenexposition und der Variabilität zwischen den Beobachtern wird diese Methode jedoch nicht häufig angewandt (9).
2.3 Computertomographie
Die Computertomographie ist in der Lage, Patienten mit ARVD zu diagnostizieren. Dery et al11 waren die ersten, die bei einem Patienten mit ARVD einen dilatierten hypokinetischen rechten Ventrikel nachwiesen.
Bei der Elektronenstrahl-Computertomographie lassen sich folgende Anzeichen für eine ARVD feststellen7:
– Vorhandensein von epikardialem Fett oder intramyokardialen Fettablagerungen
– auffällige Trabekel mit geringer Abschwächung
– dilatierter hypokinetischer rechter Ventrikel
– Scalloped-Erscheinung der rechten Ventrikelwand
Zurzeit ist die Computertomographie aufgrund der hohen Strahlenbelastung nicht das optionale bildgebende Verfahren für die Erstuntersuchung.
2.4 Kardiovaskuläre Magnetresonanztomographie
Die Magnetresonanztomographie (MR) ist ein hervorragendes Instrument zur Darstellung des rechten Ventrikels. Sie ermöglicht eine dreidimensionale Beurteilung der ventrikulären Anatomie und des Volumens und kann im Vergleich zu anderen Techniken den Ersatz von myokardialem Fett- und Fibrofettgewebe besser erkennen, obwohl eine Fettinfiltration des rechten Ventrikels nicht ausschließlich auf ARVD zurückzuführen ist, da sie bei mehr als 50 % der normalen Herzen älterer Menschen auftritt. Das Vorhandensein einer transmuralen Verfettung oder einer diffusen Ausdünnung des Myokards der rechten Herzkammer sollte jedoch als Hauptkriterium für die Diagnose einer ARVD angesehen werden.
MR kann auch zur Bewertung der systolischen und diastolischen Funktion verwendet werden. Mehrere Studien haben sich mit dem Vorhandensein einer rechtsventrikulären diastolischen Dysfunktion als frühem Marker der Erkrankung befasst (12)
Die typischen Kriterien, die mit MR nachgewiesen werden können, sind:
– Vorhandensein von Bereichen mit hoher Signalintensität, die auf die Substitution des Myokards durch Fett hinweisen (Hauptkriterium)
– faserig-fettiger Ersatz, der zu einer diffusen Ausdünnung des rechtsventrikulären Myokards führt (Hauptkriterium)
– Aneurysma des rechten Ventrikels und des rechtsventrikulären Ausflusstrakts (Hauptkriterium)
– Dilatation des rechten Ventrikels und des rechtsventrikulären Ausflusstrakts (wenn schwer, Hauptkriterium; wenn leicht, Hauptkriterium)
– regionale Kontraktionsanomalien (Nebenkriterium)
– globale systolische Dysfunktion (Hauptkriterium) und globale diastolische Dysfunktion (Nebenkriterium)
Die kardiovaskuläre Magnetresonanztomographie liefert wichtige anatomische, morphologische, funktionelle und strömungsdynamische Kriterien für die Diagnose der ARVD, obwohl die Diagnose der ARVD auf der Grundlage der Task Force-Kriterien und nicht nur auf der Grundlage struktureller Anomalien gestellt werden muss.
2.5 Endomyokardbiopsie
Die histologische Diagnose ist definitiv, die Endomyokardbiopsie ist jedoch umstritten, da die Erkrankung segmental verläuft und die Proben in der Regel aus dem Septum entnommen werden (13). Es können Komplikationen wie Tamponade und Perforation auftreten.
4 – Behandlung von Patienten mit ARVD
Vor der Behandlung müssen wir die Prädiktoren für die Mortalität und die Risikostratifizierung kennen. In der Studie von Hulot et al. (14) wurde festgestellt, dass mindestens eine Episode einer ventrikulären Tachykardie mit Linksschenkelblock, klinische Anzeichen einer rechtsventrikulären Insuffizienz und eine linksventrikuläre Dysfunktion mit kardiovaskulären Todesfällen assoziiert waren.
Das Problem ist, dass das Auftreten des plötzlichen Todes nicht mit dem Fortschreiten der Krankheit zusammenhängt und der plötzliche Tod die erste Manifestation der Krankheit sein könnte.
1.- Antiarrhythmika
Antiarrhythmika sind die erste und am häufigsten verwendete Therapie. Beta-adrenerge Blocker werden empfohlen, um adrenerge stimulierte Arrhythmien zu reduzieren.
Das erfolgreichste Medikament ist Sotalol. Sotalol war bei Patienten mit induzierbarer und nicht induzierbarer ventrikulärer Tachykardie (VT) in Dosierungen von 320 bis 480 mg/Tag wirksamer als Betablocker oder Amiodaron (Sotalol verhinderte VT während der programmierten ventrikulären Stimulation bei 68 %, während Amiodaron 26 %, Klasse Ia und Ib 5,6 % und Klasse Ic nur bei 3 % der Patienten verhinderte). (15).
2.- Katheterablation
Die Hochfrequenzablation (RF) wird bei Medikamentenrefraktärität/-intoleranz oder unaufhörlichen ventrikulären Tachykardien eingesetzt. Ziel der HF-Ablation ist die Ausschaltung der Erregungsleitungsbahnen. Sie führt nur in 30 bis 65 % der Fälle zu einem vollständigen Erfolg. Aufgrund des fortschreitenden und diffusen Charakters der Erkrankung ist es schwierig, multiple arrhythmogene Herde zu beseitigen16.
Das patologische Substrat wird bei elektrophysiologischen Untersuchungen als fraktioniertes endokardiales Signal mit geringer Amplitude aufgezeichnet, das eine verringerte lokale endokardiale Überleitungsgeschwindigkeit widerspiegelt7.
3.- Therapie mit implantierbarem Kardioverter-Defibrillator
Patienten, bei denen ein hohes Risiko für einen plötzlichen Herztod besteht, sollten einen implantierbaren Kardioverter-Defibrillator (ICD) erhalten. Es handelt sich um Patienten, die 1) nach einem Herzstillstand mit Synkope reanimiert wurden, 2) bedrohliche Herzrhythmusstörungen haben, die durch eine medikamentöse Antiarrhythmie-Therapie nicht vollständig unterdrückt werden, und 3) bei denen in der Familie ein Herzstillstand bei Verwandten ersten Grades aufgetreten ist (Primärprävention).
ICDs arbeiten mit antitachykarden Schrittmachern und Defibrillationsschocks, wenn Arrhythmien auftreten.
Die ICD-Therapie ist bei Patienten mit ARVD durchführbar und sicher und weist eine geringe Inzidenz von Kurz- und Langzeitkomplikationen auf. Mehr als drei Viertel der ARVD-Patienten erhielten während einer durchschnittlichen Nachbeobachtungszeit von 3,5 Jahren eine angemessene ICD-Therapie (17)
Es kann jedoch zu Komplikationen bei der ICD-Therapie kommen, da das Myokard des rechten Ventrikels durch Fett und fibrotisches Gewebe ersetzt wird. Dazu gehören Perforationen aufgrund der Ausdünnung der rechten Ventrikelwand, Schwierigkeiten bei der Platzierung der Elektroden aufgrund unzureichender R-Wellen-Amplituden oder hoher Stimulationsschwellen, unzureichende Erkennung oder Stimulation während der Nachbeobachtung aufgrund des Fortschreitens der Krankheit und fehlende Beendigung ventrikulärer Arrhythmien aufgrund steigender Defibrillationsschwellen (18).
4.- Behandlung der Herzinsuffizienz
Wenn eine rechtsventrikuläre oder biventrikuläre Insuffizienz auftritt, besteht die Behandlung aus der gängigen Therapie der Herzinsuffizienz einschließlich Diuretika, Betablockern, Angiotensin-konvertierenden Enzyminhibitoren und Antikoagulanzien.
Die kurative Therapie bei refraktärer kongestiver Herzinsuffizienz und/oder Arrhythmien ist die Herztransplantation.
Abbildung 1. Aufzeichnung einer Postexzitations-Epsilon-Welle (Pfeile) in den rechten präkordialen Ableitungen.
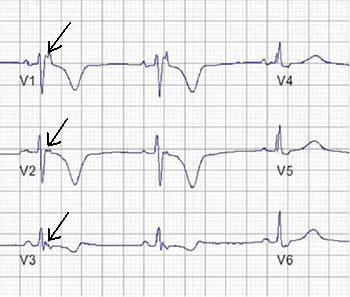
Abbildung 2. 12-Ableitungs-EKG-Aufzeichnung einer VT mit Linksschenkelblock-Morphologie
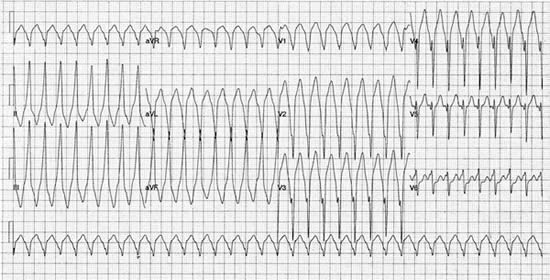
Tabelle 1. Kriterien für die Diagnose einer rechtsventrikulären DysplasieI Globale und/oder regionale Dysfunktion und strukturelle Veränderungen
| Major | Schwere Dilatation und Verringerung der rechtsventrikulären Auswurffraktion mit keiner (oder nur leichter) LV-Beeinträchtigung |
| Lokalisierte rechtsventrikuläre Aneurysmen (akinetische oder dyskinetische Bereiche mit diastolischer Ausbuchtung) | |
| Schwere segmentale Dilatation des rechten Ventrikels | |
| Mäßige globale rechtsventrikuläre Dilatation und/oder Verringerung der Auswurffraktion bei normalem linken Ventrikel | |
| Milde segmentale Dilatation des rechten Ventrikels | |
| Regionale rechtsventrikuläre Hypokinesie |
II Gewebecharakterisierung der Wände
| Major | Fibrinöser Ersatz des Myokards bei Endomyokardbiopsie |
III Repolarisationsanomalien
| Minor | Invertierte T-Wellen in den rechten präkordialen Ableitungen (V2 und V3) bei Personen im Alter von mehr als 12 J; bei fehlendem Rechtsschenkelblock |
IV Depolarisations-/Leitungsanomalien
| Major | Epsilonwellen oder lokalisierte Verlängerung (>110 ms) des QRS-Komplexes in den rechten präkordialen Ableitungen (V1-V3) |
| Minor | Spätpotentiale (signalgemitteltes EKG) |
V Arrhythmien
| Minor | Ventrikuläre Tachykardie vom Typ Linksschenkelblock (anhaltend und nicht anhaltend) (EKG, Holter, Belastungstest). |
| Häufige ventrikuläre Extrasystolen (mehr als 1000/24h) |
VI Familienanamnese
| Hauptsächlich | Familiäre Erkrankung, die bei der Nekropsie oder Operation bestätigt wurde |
| Minor | Familiäre Vorgeschichte eines vorzeitigen plötzlichen Todes (< 35 J.) aufgrund einer vermuteten rechtsventrikulären Dysplasie. |
| Familienanamnese (klinische Diagnose auf der Grundlage der derzeitigen Kriterien) |
Tabelle 2. Vorgeschlagene Änderung der Task Force für die Diagnose einer familiären ARVD
ARVD bei einem Verwandten ersten Grades plus eines der folgenden Kriterien:
| 1.- EKG | T-Wellen-Inversion in rechten präkordialen Ableitungen (V2 und V3) |
| 2.- SAECG | Spätpotentiale im Signal-Mittelwert-EKG (SAECG) |
| 3.- Arrhythmie | LBBB-Typ VT im EKG, Holter-Monitoring oder bei Belastungstests. Extrasystolen > 200 über einen Zeitraum von 24 Stunden |
| 4.- Strukturelle oder funktionelle Anomalie des RV | Milde globale RV-Dilatation und/oder EF-Reduktion bei normalem LV Milde segmentale Dilatation des RV Regionale RV-Hypokinesie |
Der Inhalt dieses Artikels spiegelt die persönliche Meinung des Autors/der Autoren wider und ist nicht unbedingt die offizielle Position der Europäischen Gesellschaft für Kardiologie
.