John J Chen, MD, PhD; Angela R McAllister, MD; Elliott H Sohn, MD
February 17, 2014
Hauptbeschwerden: Vermindertes Sehvermögen und ein zentrales Skotom auf beiden Augen (OU)
Geschichte der aktuellen Erkrankung: Der Patient ist ein 43-jähriger Mann, der sich mit einer Sehverschlechterung und einem zentralen Skotom (OU) seit 10 Jahren vorstellte, das sich zunehmend verschlechtert hat. Er beschreibt sein Sehvermögen als einen verschwommenen Fleck in der Mitte seines Gesichtsfeldes auf beiden Seiten. Der Patient war vor zwei Jahren bei einem Optiker und konnte auf beiden Augen nicht besser als 20/40 refraktiert werden. Der Patient hatte in den letzten zwei Jahren intermittierende Photopsien auf beiden Augen. Er leugnet Floater.
Augenvorgeschichte: Keine
Medizinische Vorgeschichte: Depressionen
Medikamente: Sertralin, Fischöl
Allergien: Keine
Familiäre Vorgeschichte: Keine
Soziale Vorgeschichte: Der Patient arbeitet als Küchenchef. Er raucht nicht und trinkt keinen Alkohol.
Überprüfung der Systeme: Alle negativ außer HPI
Augenuntersuchung
Sehschärfe
- Rechtes Auge (OD): 20/60
- Linkes Auge (OS): 20/60
Pupillen: 5→3, keine RAPD OU
Extraokulare Bewegungen: Volle OU
Konfrontationsgesichtsfelder: Vollständig OU
Augeninnendruck:
- OD: 21 mmHg
- OS: 19 mmHg
Extern
Lichtlampenuntersuchung
- Lider/Wimpern: Normal OU
- Bindehaut/Sklera: Normal OU
- Hornhaut: Klar OU
- Vordere Augenkammer: Tief und ruhig OU
- Iris: Normal OU
- Linse: Klar OU
- Glaskörper: Normal OU
Dilatierte Fundusuntersuchung
Die Sehnerven haben ein Verhältnis von Exkavation zu Scheibe von 0,2 OU. Die Makula beider Augen hat einen gräulichen Schimmer mit oberflächlichen Kristallen, rechtwinkligen Venolen und telangiektatischen Gefäßen, die temporal stärker ausgeprägt sind. Die Gefäße und die periphere Netzhaut sind normal (OU). Es liegt keine hintere Glaskörperabhebung vor (Abbildung 1).
Zusatzuntersuchungen
Fundusfotos zeigen einen gräulichen Schimmer mit oberflächlichen Kristallen, Venolen im rechten Winkel und telangiektatischen Gefäßen, die temporal in der Makula sowohl des rechten (A) als auch des linken (B) Auges stärker ausgeprägt sind (Abbildung 1).
Die Fluoreszeinangiographie (FA) zeigt telangiektatische Gefäße, die die Fovea umgeben und zeitlich stärker ausgeprägt sind, mit Leckage OU (Abbildung 2).
Die optische Kohärenztomographie (OCT) im Spektralbereich zeigt kleine foveale zystoide Hohlräume sowohl im rechten (A) als auch im linken (B) Auge. Die zentrale Makuladicke beträgt 331 Mikrometer OD und 320 Mikrometer OS (Abbildung 3).
Die Autofluoreszenz-Bildgebung zeigt eine leichte Zunahme der Autofluoreszenz in der Fovealregion sowohl des rechten (A) als auch des linken (B) Auges (Abbildung 4).
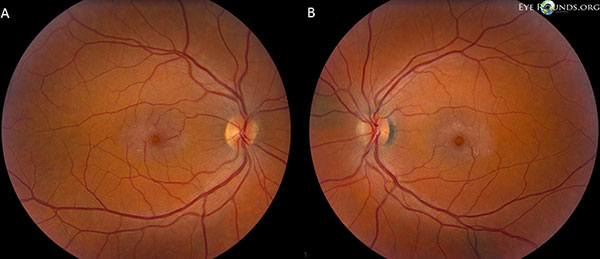
Abbildung 1. Fundusfotos zeigen einen gräulichen Schimmer mit oberflächlichen Kristallen, Venolen im rechten Winkel und telangiektatischen Gefäßen, die in der Makula sowohl des rechten (A) als auch des linken (B) Auges zeitlich stärker ausgeprägt sind.
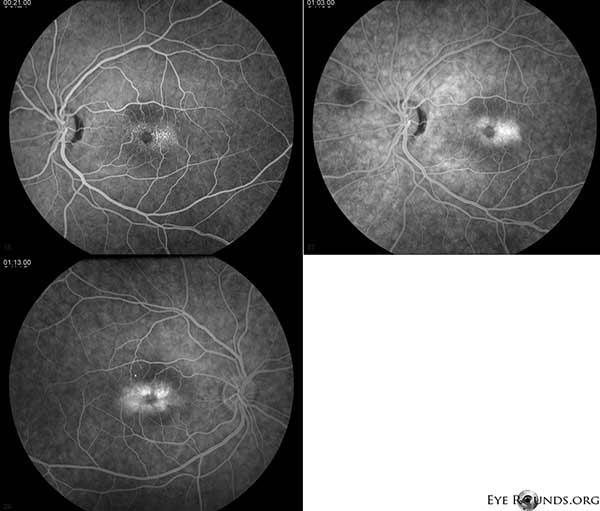
Abbildung 2. Die Fluorescein-Angiographie zeigt telangiektatische Gefäße, die die Fovea umgeben und zeitlich stärker ausgeprägt sind, mit Leckage OU.
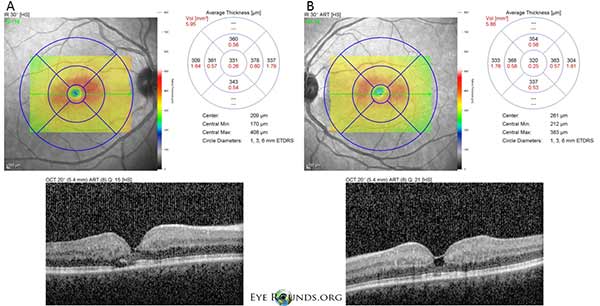
Abbildung 3: Die optische Kohärenztomographie (OCT) im Spektralbereich zeigt kleine foveale zystoide Hohlräume sowohl im rechten (A) als auch im linken (B) Auge. Die zentrale Makuladicke beträgt 331 μm OD und 320 μm OS.
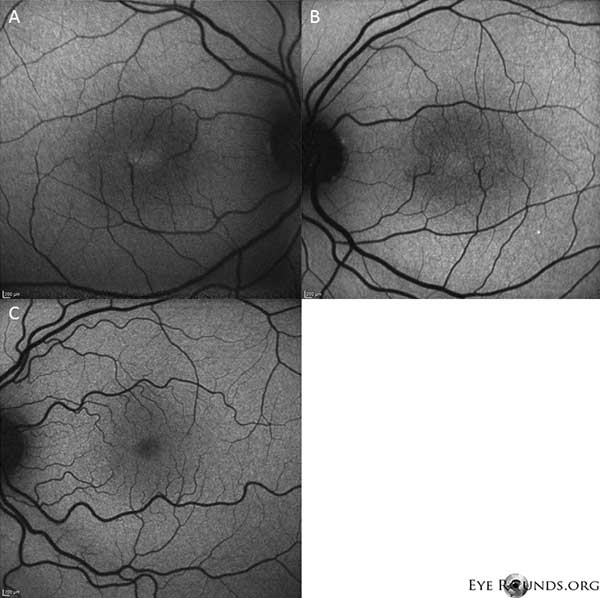
Abbildung 4: Die Autofluoreszenz-Bildgebung zeigt eine Zunahme der Autofluoreszenz in der Fovealregion sowohl des rechten (A) als auch des linken (B) Auges. Zum Vergleich wird ein Auge mit normaler Autofluoreszenz gezeigt (C).
Diagnose
Idiopathische juxtafoveale Teleangiektasie, Typ II (Makuläre Teleangiektasie Typ 2 oder Mac Tel 2)
Erörterung
Idiopathische juxtafoveale Teleangiektasie (IJFT), auch bekannt als idiopathische makuläre Teleangiektasie, ist eine seltene Erkrankung, die durch telangiektatische Gefäße in der juxtafoveolären Region eines oder beider Augen gekennzeichnet ist. Nach Gass kann die IJFT anhand des Phänotyps in drei Gruppen eingeteilt werden: Typ I ist typischerweise eine einseitige Erkrankung, die durch parafoveale Dilatation der Kapillaren, Mikroaneurysmen, Leckagen und Lipidablagerungen gekennzeichnet ist; Typ II ist die häufigste Form der IJFT und tritt typischerweise mit bilateralen juxtafovealen Teleangiektasien mit minimalem Exsudat auf; Typ III ist extrem selten und durch okklusive Teleangiektasien gekennzeichnet. Diese Übersicht konzentriert sich auf IJFT Typ II (makuläre Teleangiektasie Typ 2 oder Mac Tel 2).
Die Prävalenz von IJFT Typ II ist nicht vollständig bekannt, aber eine große Studie schätzte eine Prävalenz von 1-5 in 22.062, während eine andere Studie schätzte, dass sie in einigen Populationen bis zu 0,1 % betragen könnte. Obwohl IJFT in jedem Alter auftreten kann, liegt das Durchschnittsalter für den Ausbruch der Krankheit bei 55 Jahren. Es gibt weder eine geschlechtsspezifische noch eine rassische Prädilektion. Obwohl es einige Fallberichte über eineiige Zwillinge mit IJFT Typ II gibt, die eine genetische Komponente vermuten lassen, gibt es derzeit keine ausreichenden Beweise aus Bevölkerungsstudien, um eine genetische Assoziation zu belegen. Mehrere Studien deuten darauf hin, dass Rauchen die Krankheit verschlimmern kann.
IJFT Typ II ist eine bilaterale Erkrankung, kann aber auch asymmetrisch sein und zu Beginn der Krankheit als einseitiger Prozess auftreten. Die Patienten beschweren sich häufig über verschwommenes Sehen, Metamorphopsie oder parazentrale Skotome.
Zu den frühen Veränderungen, die bei IJFT Typ II zu beobachten sind, gehören eine parafoveale Vergrauung der Netzhaut, oberflächliche kristalline Ablagerungen, subfoveale zystoide Vertiefungen, parafoveale Teleangiektasien und rechtwinklige Gefäße. Die Sehschärfe nimmt langsam ab und ist häufig mit einer Hyperplasie des retinalen Pigmentepithels (RPE) verbunden. Bei etwa einem Drittel der Patienten kann als akute Komplikation eine tiefe retinale Neovaskularisation mit retinalen Feedern, die subretinale Neovaskularisation (SRNV), auftreten, die dann als proliferative Form bezeichnet wird. Das natürliche Fortschreiten der Krankheit führt bei der Mehrzahl der Patienten mit IJFT Typ II zu einem erheblichen Sehverlust. In einer Arbeit von Watzke et al. entwickelten 15 bzw. 20 Augen entweder eine zentrale RPE-Hyperplasie oder eine SRNV mit einer Sehschwäche von 20/70 oder schlechter im Laufe von 15 Jahren.
Fundusbefunde von IJFT Typ II auf der Biomikroskopie können subtil sein, insbesondere in der Frühphase der Erkrankung, und daher sind Bildgebung mit FA, OCT und Fundusautofluoreszenz wichtig für die Diagnosestellung. Die FA hebt die parafovealen telangiektatischen Gefäße hervor, die eine frühe Hyperfluoreszenz mit Leckage aufweisen. Diese sind häufig temporal der Fovea stärker ausgeprägt. Das OCT zeigt subfoveale zystoide Räume, normalerweise ohne zystoides Makulaödem. Bei fortgeschrittener Erkrankung zeigt das OCT eine Zerstörung der Photorezeptoren und eine Atrophie der äußeren Netzhaut. Die Befunde der Fundusautofluoreszenz sind pathognomonisch für MacTel II und zeigen einen Verlust der physiologischen Hypoautofluoreszenz – d. h. eine erhöhte Autofluoreszenz – in der Fovea.
Die Pathogenese der IJFT Typ II ist unklar, könnte aber eher mit Anomalien in den parafoveolären Müllerzellen als mit einer primären Anomalie der Netzhautkapillaren zusammenhängen. Müllerzellen sind wichtig für die Gesundheit des retinalen Kapillarendothels und der umgebenden Netzhaut. Es wurde postuliert, dass eine Funktionsstörung der Müllerzellen bei IJFT Typ II zu einer Degeneration des Endothels führt, was wiederum eine Proliferation der Netzhautkapillaren und Teleangiektasien zur Folge haben kann. Als Beleg dafür wurde in der Histopathologie von Patienten mit IJFT Typ II eine perifoveale Abreicherung von Müllerzellen festgestellt. Es wird angenommen, dass die oberflächlichen Kristalle, die bei Patienten mit IJFT Typ II zu sehen sind, die Fußplatten degenerierter Müllerzellen darstellen. Darüber hinaus wurde spekuliert, dass die auf dem OCT bei IJFT Typ II zu sehenden Zwischenräume eher einen Gewebeverlust durch Netzhautdegeneration darstellen, der speziell auf die Funktionsstörung oder den Verlust von Müllerzellen zurückzuführen ist, als flüssigkeitsgefüllte zystische Zwischenräume.
Ein besseres Verständnis des Krankheitsmechanismus bei IJFT Typ II ist wichtig, da es nach wie vor keine definitive Behandlung für den Sehverlust gibt, der bei der nicht-proliferativen Form von IJFT II auftritt. Bevacizumab hat sich bei der Behandlung der mit IJFT Typ II assoziierten SRNV als wirksam erwiesen, scheint aber den Verlauf oder die zystischen Veränderungen bei der nicht proliferativen IJFT nicht konsequent zu beeinflussen. Auch Ranibizumab konnte in einer prospektiven Interventionsstudie bei Patienten mit nichtproliferativer IJFT Typ II keinen funktionellen Nutzen nachweisen, obwohl es zu einer signifikanten Verringerung der Netzhautdicke und einer Abnahme der Leckage bei FA führte. Orale Karbonsäureanhydrasehemmer führten ebenfalls zu einer signifikanten Verringerung der Netzhautdicke, verbesserten jedoch die Sehschärfe nicht signifikant. Es wurden mehrere andere Maßnahmen erprobt, darunter ein fokaler Gitterlaser, eine photodynamische Therapie und intravitreales Triamcinolon, die jedoch weder zu einer deutlichen Verbesserung der zystoiden Hohlräume noch der Sehschärfe bei Patienten mit IJFT Typ II führten. Es ist wichtig, eine wirksame Behandlung zu finden, da die Mehrheit der Patienten mit IJFT Typ II im Laufe der Zeit eine deutliche Verschlechterung des Sehvermögens entwickelt.
Unser Patient weist alle frühen Befunde einer nicht-proliferativen IJFT Typ II auf, einschließlich einer parafovealen Vergrauung der Netzhaut, oberflächlichen kristallinen Netzhautablagerungen, Gefäßen im rechten Winkel und parafovealen Teleangiektasien (Abbildung 1). Mit der FA wurden die parafovealen Teleangiektasien weiter hervorgehoben, die deutliche Leckagen und Verfärbungen der Netzhaut aufwiesen (Abbildung 2). Das OCT zeigte charakteristische subfoveale zystoide Räume (Abbildung 3). Schließlich zeigte die Fundusautofluoreszenz eine leichte Erhöhung der fovealen Autofluoreszenz, die mit IJFT Typ II vereinbar ist (Abbildung 4). Glücklicherweise zeigte unser Patient keine Anzeichen einer weiter fortgeschrittenen Erkrankung, einschließlich keiner Anzeichen einer Hyperplasie des retinalen Pigmentepithels oder SRNV. Der Patient wurde zunächst mit PO Methazolamid 50 mg bid behandelt und zeigte innerhalb von 1,5 Monaten eine Abnahme der Makuladicke (Abbildung 5). Anschließend wurde er aus versicherungstechnischen Gründen auf PO Acetazolamid umgestellt, und im Laufe des nächsten Jahres nahmen die Makuladicke und die subfovealen Zysten weiter ab, obwohl er nur 125 mg bid vertrug (Abbildung 5). Bei der letzten Nachuntersuchung kam es auch zu einer leichten, nicht signifikanten Verbesserung der Sehschärfe auf 20/50 OD und 20/40 OS.
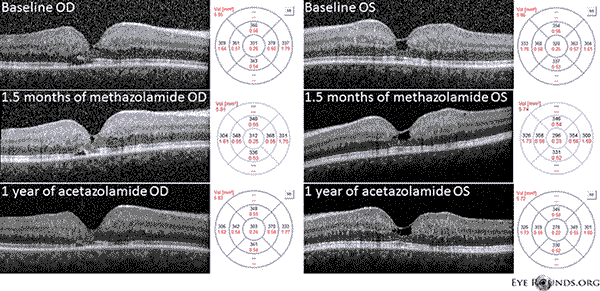
Abbildung 5: Die optische Kohärenztomographie (OCT) im Spektralbereich zeigt subfoveale zystenähnliche Räume OU bei Studienbeginn. Die Karte der Netzhautdicke ist rechts dargestellt (CMT = 331 μm OD, CMT = 320 μm OS). Nach 1,5 Monaten Methazolamid-Behandlung kam es zu einer Verringerung der Makuladicke (CMT = 312 μm OD, CMT = 296 μm OS). Nach einem Jahr Behandlung mit Acetazolamid kam es zu einer weiteren Abnahme der Zystenräume und der Makuladicke (CMT = 303 μm OD, CMT = 278 μm OS). Die Bilder wurden im selben Meridian aufgenommen und zum ursprünglichen Besuch registriert.
Differenzialdiagnose
- diabetisches Makulaödem
- pseudophakes Makulaödem
- lamellares/makuläres Loch
- Coats-Krankheit
- retinaler Venenverschluss
- Strahlenretinopathie
- Eales-Krankheit
- okulares ischämisches Syndrom
- kristalline Retinopathie
- IJFT Typ I und III (siehe Tabelle).
- Die proliferative Erkrankung kann mit einer choroidalen Neovaskularisation bei altersbedingter Makuladegeneration verwechselt werden.
Tabelle: Merkmale der drei Typen der idiopathischen juxtafoveolären Teleangiektasie
| Typen der IJFT* |
Epidemiologie |
Symptome und Symptome |
Behandlung |
Prognose |
|
IJFT Typ I |
Überwiegend männlich. Durchschnittsalter 40 Jahre. |
Einseitig prominente sichtbare telangiektatische Netzhautkapillaren mit Makulaödem und Lipidablagerungen/Exsudat. |
Laserphotokoagulation kann Exsudation reduzieren und Sehvermögen stabilisieren. |
Variabel, die Mehrheit entwickelt sich unbehandelt zu 20/70 oder schlechter |
|
IJFT Typ II |
Gleiche Geschlechtspräferenz. Durchschnittsalter 55 Jahre. |
Bilaterale parafoveale Ergrauung der Netzhaut, oberflächliche kristalline Ablagerungen, subfoveale zystoide Hohlräume, parafoveale Teleangiektasien (deutlicher bei FA), rechtwinklige Gefäße, Hyperplasie des RPE. SRNV entwickelt sich bei etwa 1/3 der Patienten. |
Keine bekannte Behandlung für nicht-proliferative IJFT Typ II. Intravitrealer Anti-VEGF für SRNV. |
Variabel, 2/3 der Augen erreichen eine Sehschärfe von 20/70 oder schlechter, verbunden mit RPE-Hyperplasie oder SRNV. |
|
IJFT Typ III |
Sehr selten |
Bilaterale perifoveale Kapillarobliteration, kapilläre Teleangiektasien und minimale Exsudation, verbunden mit systemischen oder zerebralen Erkrankungen. |
Wegen ihrer Seltenheit unbekannt |
Variabel, wegen ihrer Seltenheit meist unbekannt |
| *Die idiopathische juxtafoveale Teleangiektasie (IJFT) ist auch als idiopathische makuläre Teleangiektasie bekannt. Nach der Klassifikation der idiopathischen makulären Teleangiektasien wird IJFT Typ I als aneurysmale Teleangiektasie und IJFT Typ II als perifoveale Teleangiektasie bezeichnet. Wegen ihrer Seltenheit wurde der IJFT-Typ III aus der Klassifikation der idiopathischen makulären Teleangiektasien herausgenommen. | ||||
Epidemiologie (IJFT Typ II)
|
Symptome
|
Symptome
|
Behandlung
|
- Yannuzzi LA, Bardal AM, Freund KB, Chen KJ, Eandi CM, Blodi B. Idiopathic macular telangiectasia. Arch Ophthalmol 2006;124(4):450-60.
- Gass JD, Blodi BA. Idiopathic juxtafoveolar retinal telangiectasis. Aktualisierung der Klassifikation und Follow-up-Studie. Ophthalmology 1993;100(10):1536-46.
- Aung KZ, Wickremasinghe SS, Makeyeva G, Robman L, Guymer RH. Die Prävalenzschätzungen der makulären Teleangiektasie Typ 2: die Melbourne Collaborative Cohort Study. Retina 2010;30(3):473-8.
- Klein R, Blodi BA, Meuer SM, Myers CE, Chew EY, Klein BE. Die Prävalenz der makulären Teleangiektasie Typ 2 in der Beaver Dam Eye Study. Am J Ophthalmol 2010;150(1):55-62 e2.
- Watzke RC, Klein ML, Folk JC, Farmer SG, Munsen RS, Champfer RJ, Sletten KR. Langfristige juxtafoveale retinale Teleangiektasien. Retina 2005;25(6):727-35.
- Nowilaty SR, Al-Shamsi HN, Al-Khars W. Idiopathic juxtafoveolar retinal telangiectasis: a current review. Middle East Afr J Ophthalmol 2010;17(3):224-41.
- Cohen SM, Cohen ML, El-Jabali F, Pautler SE. Optische Kohärenztomographie-Befunde bei nichtproliferativer idiopathischer juxtafovealer retinaler Teleangiektasie der Gruppe 2a. Retina 2007;27(1):59-66.
- Schmitz-Valckenberg S, Fan K, Nugent A, Rubin GS, Peto T, Tufail A, Egan C, Bird AC, Fitzke FW. Korrelation von funktionellen Beeinträchtigungen und morphologischen Veränderungen bei Patienten mit idiopathischer juxtafovealer retinaler Teleangiektasie der Gruppe 2A. Arch Ophthalmol 2008;126(3):330-5.
- Wong WT, Forooghian F, Majumdar Z, Bonner RF, Cunningham D, Chew EY. Fundusautofluoreszenz bei idiopathischer makulärer Teleangiektasie Typ 2: Korrelation mit optischer Kohärenztomographie und Mikroperimetrie. Am J Ophthalmol 2009;148(4):573-83.
- Gass JD. Histopathologische Untersuchung einer mutmaßlichen parafovealen Teleangiektasie. Retina 2000;20(2):226-7.
- Tout S, Chan-Ling T, Hollander H, Stone J. The role of Muller cells in the formation of the blood-retinal barrier. Neuroscience 1993;55(1):291-301.
- Newman E, Reichenbach A. The Muller cell: a functional element of the retina. Trends Neurosci 1996;19(8):307-12.
- Powner MB, Gillies MC, Tretiach M, Scott A, Guymer RH, Hageman GS, Fruttiger M. Perifoveal muller cell depletion in a case of macular telangiectasia type 2. Ophthalmology 2010;117(12):2407-16.
- Gass JDM. Stereoskopischer Atlas der Makulaerkrankungen : Diagnose und Behandlung. 4th ed. St. Louis: Mosby, 1997.
- Roller AB, Folk JC, Patel NM, Boldt HC, Russell SR, Abramoff MD, Mahajan VB. Intravitreales Bevacizumab zur Behandlung der proliferativen und nicht-proliferativen idiopathischen makulären Teleangiektasie Typ 2. Retina 2011;31(9):1848-55.
- Mandal S, Venkatesh P, Abbas Z, Vohra R, Garg S. Intravitreal bevacizumab (Avastin) for subretinal neovascularization secondary to type 2A idiopathic juxtafoveal telangiectasia. Graefes Arch Clin Exp Ophthalmol 2007;245(12):1825-9.
- Konstantinidis L, Mantel I, Zografos L, Ambresin A. Intravitreales Ranibizumab als primäre Behandlung für neovaskuläre Membranen in Verbindung mit idiopathischer juxtafovealer retinaler Teleangiektasie. Graefes Arch Clin Exp Ophthalmol 2009;247(11):1567-9.
- Matsumoto Y, Yuzawa M. Intravitreal bevacizumab therapy for idiopathic macular telangiectasia. Jpn J Ophthalmol 2010;54(4):320-4.
- Kovach JL, Rosenfeld PJ. Bevacizumab (Avastin) Therapie für idiopathische makuläre Teleangiektasie Typ II. Retina 2009;29(1):27-32.
- Charbel Issa P, Finger RP, Kruse K, Baumuller S, Scholl HP, Holz FG. Monatliche Ranibizumab für nicht-proliferative makuläre Teleangiektasie Typ 2: eine 12-monatige prospektive Studie. Am J Ophthalmol 2011;151(5):876-886 e1.
- Chen JJ, Sohn EH, Folk JC, Mahajan VB, Kay CN, Boldt HC, Russell SR. Verminderte Makuladicke bei nichtproliferativer makulärer Telangiektasie Typ 2 mit oralen Carbonic Anhydrase Inhibitoren. Retina IN PRESS; 2014.
- Park DW, Schatz H, McDonald HR, Johnson RN. Grid-Laser-Photokoagulation für Makulaödeme bei bilateraler juxtafovealer Teleangiektasie. Ophthalmology 1997;104(11):1838-46.
- De Lahitte GD, Cohen SY, Gaudric A. Lack of apparent short-term benefit of photodynamic therapy in bilateral, acquired, parafoveal telangiectasis without subretinal neovascularization. Am J Ophthalmol 2004;138(5):892-4.
- Wu L, Evans T, Arevalo JF, Berrocal MH, Rodriguez FJ, Hsu M, Sanchez JG. Langfristige Wirkung von intravitrealem Triamcinolon im nicht-proliferativen Stadium der idiopathischen parafovealen Teleangiektasie vom Typ II. Retina 2008;28(2):314-9.