Abbildung 1. Toll-like-Rezeptor-Signalweg
Was ist ein Toll-like-Rezeptor?
Toll-like-Rezeptoren (TLRs) sind eine Klasse von Proteinen, die eine Schlüsselrolle in der angeborenen Immunität spielen. Es handelt sich um transmembranöse Einzeldomänenrezeptoren, die zu den Mustererkennungsrezeptoren (PRRs) gehören, die normalerweise in Sentinel-Zellen wie Makrophagen, dendritischen Zellen und vielen anderen nicht-immunen Zellen wie Fibroblasten und Epithelzellen exprimiert werden. Sie erkennen strukturell konservierte, von Mikroben stammende Moleküle, die als pathogen-assoziierte molekulare Muster (PAMPs) bezeichnet werden, oder von geschädigten Zellen selbst stammende Moleküle, die als schädigungsassoziierte molekulare Muster (DAMPs) bezeichnet werden. Zu den PAMPs gehören verschiedene bakterielle Zellwandkomponenten wie Lipopolysaccharid (LPS), Peptidoglykan (PGN) und Lipopeptide sowie Flagellin, bakterielle DNA und virale doppelsträngige RNA. Zu den DAMPs gehören intrazelluläre Proteine wie Hitzeschockproteine sowie Proteinfragmente aus der extrazellulären Matrix. PRRs aktivieren nachgeschaltete Signalwege, die zur Induktion von angeborenen Immunantworten führen, indem sie entzündliche Zytokine, Typ-I-Interferon (IFN) und andere Mediatoren produzieren. Diese Prozesse lösen nicht nur unmittelbare Abwehrreaktionen des Wirts aus, wie z. B. Entzündungen, sondern lösen auch antigenspezifische adaptive Immunreaktionen aus und steuern diese. Diese Reaktionen sind sowohl für die Beseitigung infizierender Mikroben als auch für die anschließende Anweisung antigenspezifischer adaptiver Immunantworten von entscheidender Bedeutung.
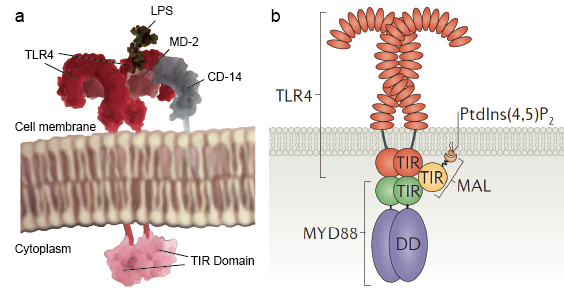
Abbildung 2. Schematische Darstellung der molekularen Struktur der TLRs.
Toll-like receptor family
Die TLR-Familie umfasst 10 Mitglieder (TLR1-TLR10) beim Menschen und 12 (TLR1-TLR9, TLR11-TLR13) bei der Maus. TLRs lokalisieren sich an der Zelloberfläche oder in intrazellulären Kompartimenten wie dem ER, Endosom und Lysosom. Zu den Zelloberflächen-TLRs gehören TLR1, TLR2, TLR4, TLR5, TLR6 und TLR10, während die intrazellulären TLRs im Endosom lokalisiert sind und TLR3, TLR7, TLR8, TLR9, TLR11, TLR12 und TLR13 umfassen (Abbildung 1). Zelloberflächen-TLRs erkennen hauptsächlich mikrobielle Membranbestandteile wie Lipide, Lipoproteine und Proteine. Intrazelluläre TLRs erkennen Nukleinsäuren, die von Bakterien und Viren stammen, und erkennen auch eigene Nukleinsäuren bei Krankheiten wie Autoimmunität.
Die Funktion des Toll-like-Rezeptors basiert in der Regel auf einem Dimerisierungsprozess zweier TLR-Moleküle, aber nicht immer. Zum Beispiel binden sich TLR-1 und TLR-2 aneinander, um einen Dimmer zu bilden, wenn sie PAMPs-Moleküle erkennen, zu denen hauptsächlich Lipoproteine, Peptidoglykane, Lipotechosäuren (LTA, Gram-), Zymosan, Mannan und tGPI-Mucin gehören. TLR-2 kann auch einen Dimmer mit TLR-6 bilden, wenn sie die gleichen oben aufgeführten PAMPs erkennen. TLR-4 kann Lipopolysaccharid (LPS, Gram+) erkennen und ein Homodimer mit einem anderen TLR-4-Molekül bilden. TLR-5 kann bakterielles Flagellin erkennen, aber sie bilden kein Dimer. TLR-11 ist bei Mäusen funktionsfähig und erkennt hauptsächlich uropathogene Bakterien. TLR-3, 7, 8, 9, 13 werden auf der Endosomenoberfläche im Zytoplasma exprimiert. TLR3 erkennt virale doppelsträngige RNA (dsRNA), kleine interferierende RNAs und Selbst-RNAs, die von geschädigten Zellen stammen. TLR-7 wird vorwiegend in plasmazytoiden DCs (pDCs) exprimiert und erkennt einzelsträngige (ss) RNA aus Viren. Er erkennt auch RNA von Streptokokken-B-Bakterien in konventionellen DCs (cDCs). TLR8 reagiert auf virale und bakterielle RNA. TLR-9 erkennt bakterielle und virale DNA, die reich an unmethylierten CpG-DNA-Motiven ist. TLR13 erkennt bakterielle 23S rRNA und unbekannte Komponenten des vesikulären Stomatitisvirus.
Obwohl es so viele Arten von TLR-Molekülen gibt, die ein breites Spektrum von Liganden erkennen, haben alle diese TLRs ein gemeinsames strukturelles Gerüst in ihren extrazellulären, ligandenbindenden Domänen. Diese Domänen weisen alle hufeisenförmige Strukturen auf, die aus leucinreichen Wiederholungsmotiven bestehen. Typischerweise bilden zwei extrazelluläre Domänen bei der Ligandenbindung ein “m“-förmiges Dimer, das das Ligandenmolekül einschließt und die transmembranen und zytoplasmatischen Domänen in enge Nähe bringt und eine nachgeschaltete Signalkaskade auslöst (Abbildung 2).
Toll-like Rezeptor-Signalweg
1. Toll-like-Rezeptor-Signalkaskade
Toll-like-Rezeptoren ermöglichen Sentinel-Zellen wie Makrophagen, Mikroben durch PAMPs wie LPS zu erkennen. LPS ist ein Bestandteil der bakteriellen Zellwand. Der Mechanismus der Erkennung von Lipopolysaccharid durch Toll-like-Rezeptoren ist komplex und erfordert mehrere zusätzliche Proteine. Ein Serumprotein, das LPS-bindende Protein, bindet LPS-Monomere und überträgt sie auf ein Protein namens CD14. CD14 kann löslich sein oder über einen Glykosylphosphatidylinositol-Anker an die Zelloberfläche binden. CD14 liefert und lädt LPS an die extrazelluläre Domäne der Toll-like-Rezeptoren. TLRs sind in der Lage, LPS mit Hilfe eines Zusatzproteins namens MD-2 zu erkennen. Wenn LPS an den Komplex aus TLR-CD14-MD2 bindet, wird die Homodimerisierung der TLRs ausgelöst. Die Konformationsänderung der extrazellulären Domänen leitet die Dimerisierung der zytoplasmatischen Toll-IL-1-Rezeptordomäne (TIR) ein. Die Konformationsänderung der TIR-Domäne bietet ein neues Gerüst, das die Rekrutierung von Adaptorproteinen zur Bildung eines Post-Rezeptor-Signalkomplexes ermöglicht. Die TIR-Domäne enthält das Adaptorprotein Myeloid Differentiation Primary-Response Protein 88 (MyD88).
MyD88 fungiert als Adaptor, der TLRs/IL-1Rs mit nachgeschalteten Signalmolekülen verbindet, die DDs haben. Es erkennt die Konformationsänderung in der TIR-Domäne der TLRs, bindet an den neuen Rezeptorkomplex und überträgt die Signalisierung durch die Interaktion der aminoterminalen (N)-terminalen Todesdomäne (DD) mit IL-1R-assoziierten Kinasen (IRAKs). Dadurch entsteht eine komplexe Signalkaskade, die die Zelle vor einer Invasion des Pathogens warnt. Es gibt 4 IRAKs (IRAK 1, 2, 4, M). Sie enthalten eine N-terminale DD und eine zentrale Serin/Threonin-Kinase-Domäne. IRAK1 und IRAK4 haben eine intrinsische Kinaseaktivität, während IRAK2 und IRAK-M keine nachweisbare Kinaseaktivität haben. IRAK4 wird durch MyD88 aktiviert und setzt die Aktivierung von IRAK1 fort. IRAK1 aktiviert dann das nachgeschaltete TRAF6. TRAF6 ist ein Mitglied der Familie der Tumornekrosefaktor-Rezeptor (TNFR)-assoziierten Faktoren (TRAF), die Zytokin-Signalübertragungswege vermitteln. Nach einer Stimulation wird TRAF6 zum Rezeptorkomplex rekrutiert und durch IRAK-1 aktiviert, das an die TRAF-Domäne von TRAF6 bindet. Anschließend dissoziiert der IRAK-1/TRAF6-Komplex vom Rezeptor und assoziiert mit der TGF-beta-aktivierten Kinase 1 (TAK1) und den TAK1-bindenden Proteinen TAB1 und TAB2. Der Komplex aus TRAF6, TAK1, TAB1 und TAB2 bewegt sich in das Zytoplasma, wo er einen großen Komplex mit anderen Proteinen, wie den E2-Ligasen Ubc13 und Uev1A, bildet. Der Komplex aus Ubc13 und Uev1A katalysiert nachweislich die Synthese einer Lys-63-verknüpften Polyubiquitin-Kette von TRAF6 und induziert dadurch die TRAF6-vermittelte Aktivierung von TAK1 und schließlich von NF-kB. Dieser oben beschriebene Signalweg wird als MyD88-abhängiger Weg bezeichnet, da das Signal von dem MyD88-Molekül ausgeht. Es gibt auch einen anderen Signalweg, der als MyD88-unabhängiger Signalweg bezeichnet wird und bei dem die Signalisierung nicht von MyD88 ausgeht. Stattdessen geht das Signal vom TRIF-Protein aus. TRIF interagiert mit TRAF6 und TRAF3.TRAF6 rekrutiert die Kinase RIP-1, die wiederum mit dem TAK1-Komplex interagiert und diesen aktiviert, was zur Aktivierung von NF-kB und MAPKs und zur Induktion von Entzündungszytokinen führt. Im Gegensatz dazu rekrutiert TRAF3 die IKK-verwandten Kinasen TBK1 und IKKi zusammen mit NEMO, um IRF3 zu phosphorylieren und zu aktivieren. IRF3 bildet ein Dimer und transloziert aus dem Zytoplasma in den Zellkern, wo es die Expression von Typ-I-IFN induziert.
2. Downstream-Signalisierung
TLRs signalisieren hauptsächlich durch die Rekrutierung spezifischer Adaptormoleküle, was zur Aktivierung der Transkriptionsfaktoren NF-kB und IRFs führt, die das Ergebnis der angeborenen Immunantworten bestimmen. Dieser nachgeschaltete Signalweg aktiviert also den Transkriptionsfaktor IRFs, den NF-kB-Signalweg und den MAKP-Signalweg. Ausführlichere Informationen über den NF-kB- und den MAKP-Signalweg finden Sie unter:
NF-kB-Signalweg, P38-Signalweg und MAKP-Signalweg.
3. Regulierung des Signalwegs
Natürlich gibt es eine negative Regulierung durch eine Reihe von Molekülen über verschiedene Mechanismen, um übermäßige Immunreaktionen zu verhindern oder zu beenden, die zu schädlichen Folgen im Zusammenhang mit Autoimmunität und entzündlichen Erkrankungen führen. Die Aktivierung des MyD88-abhängigen Signalwegs wird durch ST2825, SOCS1 und Cbl-b unterdrückt, und die Aktivierung des TRIF-abhängigen Signalwegs wird durch SARM und TAG unterdrückt. Diese Moleküle assoziieren mit MyD88 oder TRIF, um sie an der Bindung an TLRs oder nachgeschaltete Moleküle zu hindern. Die Aktivierung von TRAF3 wird durch SOCS3 und DUBA negativ reguliert. TRAF6 wird durch eine Reihe von Hemmstoffmolekülen wie A20, USP4, CYLD, TANK, TRIM38 und SHP gehemmt. Die Aktivierung von TAK1 wird durch TRIM30a und A20 gehemmt.
4. Zusammenhang mit Krankheiten
Da TLR an der LPS-Sensierung beteiligt ist und eine Rolle bei der Sepsis spielen könnte, ist die gezielte Beeinflussung von TLRs für die Behandlung verschiedener Krankheiten wichtig. Neben der Beeinflussung von TLR-Reaktionen zur Behandlung von Infektionen durch Krankheitserreger besteht eine offensichtliche klinische Anwendung der aus TLR-Studien gewonnenen Erkenntnisse darin, TLR-Liganden als Impfstoffadjuvantien einzusetzen. Darüber hinaus wurde in der Klinik auch eine TLR-Hemmung versucht, deren Ziel es ist, übermäßige Entzündungen zu begrenzen, die vermutlich durch die Überaktivierung eines bestimmten TLR ausgelöst werden.
| Takeda, Kiyoshi, und Shizuo Akira. „TLR signaling pathways.“ Seminars in Immunology. Vol. 16. No. 1. Academic Press, 2004. | |
| Akira, Shizuo, und Kiyoshi Takeda. „Toll-like receptor signalling“. Nature Reviews Immunology 4.7 (2004): 499-511. | |
| Lim, Kian-Huat, und Louis M. Staudt. „Toll-like receptor signaling.“ Cold Spring Harbor perspectives in biology 5.1 (2013): a011247. | |
| undTaro Kawai, Takumi Kawasaki. „Toll-like receptor signaling pathways“. Pattern Recognition Receptors and Cancer 8916 (2015): 7. | |
| Gay, Nicholas J., et al. „Assembly and localization of Toll-like receptor signaling complexes.“ Nature Reviews Immunology 14.8 (2014): 546-558. | |
| O’Neill, Luke AJ, Douglas Golenbock, and Andrew G. Bowie. „Die Geschichte der Toll-like-Rezeptoren, die die angeborene Immunität neu definieren“. Nature Reviews Immunology 13.6 (2013): 453-460. | |
| Botos, Istvan, David M. Segal, and David R. Davies. „The structural biology of Toll-like receptors“. Structure 19.4 (2011): 447-459. |