Dieses Kapitel soll dem Studenten einen Einblick in die Analysemethoden geben, die bei der technischen Untersuchung von Eisen und Stahl verwendet werden. Die Natur dieser Arbeit verhindert eine detaillierte Behandlung des Themas – eine Behandlung, die eine Schätzung einer oder mehrerer der folgenden Substanzen erfordern kann: Kohlenstoff (frei und gebunden), Schwefel, Silizium, Phosphor, Mangan, Titan, Kupfer, Nickel, Kobalt, Chrom, Aluminium, Arsen, Antimon, Zinn, Wolfram, Vanadium, Stickstoff, Eisen. Im Allgemeinen werden die meisten Schätzungen für Kohlenstoff, Schwefel, Silizium und Phosphor benötigt. Von den anderen genannten Elementen und Verbindungen sind bei Spezialstählen die Bestimmungen eines oder mehrerer Elemente erforderlich. Für Informationen zu diesen Bestimmungen wird der Student auf Chemical Analysis and Foundry Chemistry, von Crobaugh; The. Chemical Analysis of Iron, von Blair; „Carbon in Steel by Direct Combustion,“ von Blount, in The Analyst, Jan. 1902; „Sulphur in Wrought Iron and Steel,“ von Auchy, in der Jour. Amer. Chem. Soc., März 1901, und andere Artikel in denselben Zeitschriften. Der Student, der weiter gehen möchte, sollte sich, wenn möglich, Zugang zu den Abhandlungen und Artikeln von Campbell, Drown und anderen verschaffen, die von Zeit zu Zeit in den verschiedenen chemischen und metallurgischen Fachzeitschriften veröffentlicht werden.
Da die Zeit des Studenten begrenzt ist, kann er die Abschätzung von Silizium und Phosphor vorerst zurückstellen, obwohl diese wegen ihrer Bedeutung sowohl für den Metallurgen als auch für den Gießer angegeben werden.
Damit der Student ein gründlicheres Verständnis des Themas erlangen kann, sind einige Anmerkungen über die Zusammensetzung und die Eigenschaften der betrachteten Substanzen nicht fehl am Platz. Zum Einfluss der verschiedenen Elemente auf den Stahl siehe The Manufacture and Properties of Structural Steel, von H. H. Campbell.
Kohlenstoff kommt im Eisen in drei Zuständen vor: graphitisch, gelöst und kombiniert. Neben diesen sind weitere Formen unter dem Mikroskop identifiziert worden.
Schwefel kommt im Eisen hauptsächlich als Sulfid FeS vor, das im geschmolzenen Eisen löslich ist.
Phosphor kommt als Eisenphosphid vor, das im geschmolzenen Eisen vollständig löslich ist.
Silizium bildet Eisensilizid, das ebenfalls im geschmolzenen Eisen löslich ist.
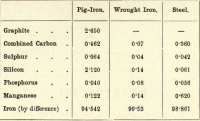
Von diesen vier Elementen ist Kohlenstoff also das einzige, das in freiem Zustand existieren kann. Die Schwankungen in den Anteilen der verschiedenen Elemente sind fast unendlich, aber die folgende kurze Tabelle gibt die ungefähre Zusammensetzung von Roheisen, Schmiedeeisen und Stahl an, obwohl jedes dieser Elemente erheblichen Schwankungen unterliegt.
Der Schüler soll Folgendes schätzen:
(1) Kohlenstoff,
(a) insgesamt.
(b) graphitisch.
(c) kombiniert.
(2) Schwefel.
(3) Silizium. (Wenn es die Zeit erlaubt.)
(4) Phosphor. (Wenn es die Zeit erlaubt.)
Kohlenstoff insgesamt
Bei dieser Schätzung wird der Kohlenstoff in CO2 umgewandelt, das in Kalilauge absorbiert wird. Aus dem Gewicht des so erhaltenen CO2 wird der Kohlenstoff berechnet.
Auf den ersten Blick scheint es das einfachste Verfahren zu sein, die Eisen- oder Stahlbohrungen direkt in einem Sauerstoffstrom zu entzünden und das so gebildete CO2 in KHO zu absorbieren. Leider hat sich diese Methode bisher entweder als ungenau erwiesen, oder dort, wo eine vollständige Verbrennung erreicht wurde, war der Apparat, der notwendig war, um der hohen Temperatur oder anderen Schwankungen in der Behandlung standzuhalten, für die technische Arbeit nicht geeignet (siehe Artikel von Blount in The Analyst). Der Student wird feststellen, dass die hier angegebene Methode vom technischen Standpunkt aus gesehen keineswegs ideal ist, was die Bequemlichkeit und Schnelligkeit betrifft, und es scheint wahrscheinlich, dass sie in naher Zukunft durch eine schnellere Methode der direkten Oxidation ersetzt wird.
Angewandte Methode: In den zahlreichen Werken zu diesem Thema findet man eine große Vielfalt von Methoden. Die hier angegebene Methode wird bei normaler Sorgfalt genaue Ergebnisse liefern. Kurz gefasst lautet sie wie folgt:-
Das Eisen wird in einer Lösung von Kalium- und Kupferdoppelchlorid gelöst, die mit HCl angesäuert wird. Das metallische Kupfer wird ausgefällt und wieder aufgelöst; das Eisen wird aufgelöst, wobei der Kohlenstoff in Suspension bleibt. Dann wird es aufgefangen und im Verbrennungsofen mit Sauerstoff entzündet und das entstandene CO2 gewogen.
Lösung des Eisens: 1 g Roheisenbohrungen abwiegen. In ein 300 c.c. Becherglas überführen. Füge 100 c.cs. CuCl2,2KCl,2H2O Lösung, die wie folgt hergestellt wird. 149,1 Teile KCl und 170,3 Teile kristallisiertes CuCl2,2H2O in Wasser auflösen. Man verdampft und kristallisiert das Doppelchlorid aus. 300 g des Doppelsalzes in destilliertem Wasser auflösen. Durch angezündeten Asbest filtrieren und in Glasflaschen mit Stopfen aufbewahren.
Zum Inhalt des Becherglases 7 c.cs. HCl zu, um die Lösung sauer zu machen. Ununterbrochen umrühren, bis sich das Eisen gelöst hat. Stelle das Becherglas mit dem Inhalt gegen Ende der Lösung auf ein Wasserbad mit einer Temperatur von etwa 60° C. Die folgenden Reaktionen laufen ab: Fe + CuCl2 = FeCl2 + Cu und Cu + CuCl2 = 2CuCl. Das KCl unterstützt lediglich die Lösung des ausgefallenen Kupfers. Etwa 40 Minuten nach der Zugabe der Doppelchloride sollte die Lösung fast vollständig sein und der größte Teil des Kupfers aufgelöst sein. Wasche die Seiten des Becherglases mit ein wenig angesäuertem Doppelchlorid ab. Der Lösung ein wenig angezündeten Asbest hinzufügen, um die kohlenstoffhaltigen Stoffe abzusetzen und zu verhindern, dass sie den Filter verstopfen (wie von Barba empfohlen).
Für die Filtration sind spezielle Platinschiffchen, die nach dem Prinzip des Gooch-Tiegels ausgestattet sind, sehr praktisch. Der Schüler kann aber auch mit Hilfe eines Gooch-Tiegels, der durch einen künstlichen Sog unterstützt wird, den Kohlenstoff abfiltrieren, wobei der Kohlenstoff mit einem Wasserstrahl ausgewaschen wird, nachdem die Flüssigkeit durch den Filter gelaufen ist. Die Kohle auf dem Filter wird vorsichtig mit heißem Wasser ausgewaschen. Der Tiegel und der Inhalt werden im Luftofen bei 100 C getrocknet. Die Kohlenstoffmasse ist nun zur Entzündung bereit.
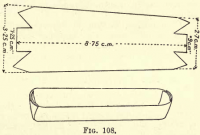
Oxidation des Kohlenstoffs: Man bereitet ein Platinschiffchen vor, indem man ein Stück Platinfolie gemäß Abb. 108 zuschneidet und die Seiten und Enden zu einer Wanne zusammenlegt. Die kohlenstoffhaltigen Stoffe und der Asbest werden aus dem Gooch in das Boot gegeben.
Der Verbrennungsofen, das Zubehör und die Armaturen müssen in Ordnung gebracht werden. Der Sauerstoffreinigungsapparat wird wieder verwendet, ist aber diesmal mit einem Dreiwegerohr versehen, dessen Hähne zwischen dem Speicher und den Reinigern eingesetzt sind. Dadurch kann ein Luftstrom durch den Apparat gezogen werden. Das Verbrennungsrohr kann aus hartem Jenaer Glas, Porzellan oder Platin bestehen. Zwischen dem Ofen und den Kalikolben werden zwei U-Rohre verwendet. Der dem Ofen zugewandte Schenkel des ersten Rohrs enthält wasserfreies CuSO4, der andere Schenkel wasserfreies CuCl. Das zweite U-Rohr enthält getrocknetes CaCl2. Diese beiden Rohre bilden den „Reinigungsstrang“. Das CuCl absorbiert jegliches Cl und die anderen Substanzen jegliches H2O. Dieses Set ist für viele Bestimmungen geeignet. Die Kalibirnen und Schutzrohre folgen, und ein Sauger sollte griffbereit sein, um bei Bedarf einen Luftstrom durch die Apparatur zu ziehen. Die Kalibirnen werden mit 8E. KHO, und das Schutzrohr mit CaCl2. Die Prüfung des Ofens und der Glühbirnen erfolgt wie zuvor beschrieben (siehe Kohle und Koks), wobei das Rohr wie in der Skizze beschickt wird und das Schiffchen vorläufig im Luftofen bei 100° C gehalten wird.
Wenn alles fertig ist und die Brenner einige Zeit ausgeschaltet waren, wird das Schiffchen mit dem Inhalt eingesetzt. Die Brenner werden vom vorderen Ende her angezündet, man arbeitet sich allmählich rückwärts und schaltet einen langsamen Sauerstoffstrom von etwa zwei Blasen pro Sekunde ein, bis die Röhre mit Sauerstoff gefüllt ist. Man regelt die Temperatur, bis das Schiffchen mattrot ist, und wenn die Lösung in den Glühbirnen Anzeichen zeigt, dass sie zum Ofen zurückläuft, erhöht man den Sauerstoffstrom auf drei oder vier Blasen pro Sekunde.
Vom Zeitpunkt des Einsetzens des Schiffchens an reichen etwa fünfzig Minuten für eine vollständige Verbrennung aus. Man schaltet den Sauerstoff ab und lässt zehn Minuten lang Luft durchströmen.
Die Kalikugeln und das Schutzrohr werden nun entfernt und gewogen, und der Kohlenstoff wird wie üblich berechnet.
(b) Graphitkohlenstoff – Das Eisen wird von den einen in HCl, von den anderen in HNO3 aufgelöst, wobei der Graphitkohlenstoff als Rückstand zurückbleibt. Für Roheisen liefern beide Methoden bei entsprechender Sorgfalt gute Ergebnisse, aber für graphithaltige Stähle empfiehlt Blair die Lösung in Salpetersäure. (Für diese Methode konsultieren Sie Blair.)
Wiegen Sie 5 g Roheisenbohrungen ab. Lösen Sie sie in 50 c.cs. SE. HCl durch leichte Hitze. Einige Minuten lang kochen lassen. Verdünnen auf 100 c.cs. (fast). Durch einen Gooch-Tiegel filtrieren. Gut mit heißem Wasser und dann mit kochendem E. KHO waschen. (Dies löst jegliches SiO2 auf.) Erneut mit heißem Wasser waschen, um das KHO zu entfernen. Tiegel und Inhalt trocknen.
Den Kohlenstoff wie zuvor durch Verbrennung bestimmen und den Prozentsatz wie üblich berechnen.
(c) Kombinierter Kohlenstoff (durch Differenz): Da der Gesamtkohlenstoff und der graphitische Kohlenstoff bekannt sind, erhält man den kombinierten Kohlenstoff durch Subtraktion des graphitischen vom Gesamtkohlenstoff.
Für direkte Methoden der Schätzung konsultieren Sie die genannten Stellen.
SCHWEFELBESTIMMUNG IN EISEN &STAHL
Es bestehen beträchtliche Meinungsverschiedenheiten über die beste Methode zur Schätzung des Schwefels in Eisen und Stahl. Die alte Methode der Königswasserlösung und BaCl2-Fällung ist zugegebenermaßen sehr ungenau; aber eine langsame Lösung in HNO3 mit sehr wenig oder gar keinem HCl, gefolgt von einer sorgfältigen Fällung durch BaCl2 in Gegenwart eines bestimmten Überschusses an HCl und mit der nötigen Sorgfalt hinsichtlich der Zeit und der Bedingungen der Fällung sowie Vorsichtsmaßnahmen gegen die Verunreinigung des Niederschlags durch Eisen – mit dieser Methode und Sorgfalt lassen sich gute Ergebnisse erzielen. Blair hingegen empfiehlt die Lösung in HCl, wobei das S als H2S entsteht, das in einer (alkalischen) Lösung von Pb(NO3)2 absorbiert wird und PbS bildet, das in HCl + KClO3 gelöst wird, wobei das S als BaSO4 ausgefällt wird. Für weitere Methoden siehe Blair, Stillman, Auchy, Crobaugh und Drown. Eine weitere gebräuchliche Methode ist die Entwicklung des S als H2S, gefolgt von der Absorption in Cadmiumchloridlösung. Das ausgefällte Cadmiumsulfid wird in HCl aufgelöst und der S durch Titration mit einer Iodlösung geschätzt, oder, was noch gebräuchlicher ist, das H2S wird in Br.-Wasser absorbiert und dann als BaSO4 ausgefällt oder in NaOH absorbiert und mit Iod titriert; letzteres ist die bevorzugte Methode. (Siehe Blair.) Die folgende Methode wird hier angegeben:-
Oxidation durch HNO3 (die sogenannte Aqua-Regia-Methode).-5 g Bohrspäne abwiegen und in ein tiefes 200 c.c.-Becherglas geben. Füge vorsichtig etwa 40 c.cs. 16E. HNO3 in Mengen von jeweils etwa 10 c.cs. zugeben, dabei das Becherglas mit einem großen Uhrglas abdecken und darauf achten, dass die Reaktion nicht zu heftig ist. Wenn der Vorgang scheinbar zum Stillstand gekommen ist, stellt man fest, ob alle Teilchen (außer dem Kohlenstoff) aufgelöst sind. Ist dies nicht der Fall, erhitzt man auf dem Sandbad und gibt 3 oder 4 Tropfen 16E. HCl zu und erwärme es, bis es sich aufgelöst hat.
Wenn die Lösung vollständig ist, füge etwas Na2CO3 hinzu, um die H2SO4 in Na2SO4 umzuwandeln, das sich beim Verdampfen nicht verflüchtigt.
Entferne das Sandbad und füge 5 c.c. starke HCl hinzu, mehr als nötig ist, um die durch das Na2CO3 ausgefallenen Eisenverbindungen aufzulösen. Das SiO2 abfiltern und C. gut waschen. Bis zur Trockne eindampfen, um das SiO2 unlöslich zu machen. Mit HCl aufnehmen und eindampfen, bis Fe2Cl6 auszukristallisieren beginnt. Dann 5 c.cs. HCl zu und filtriert, falls ein Rückstand vorhanden ist. (Wenn kein Rückstand vorhanden ist, war kein SiO2 in Lösung, und das Eindampfen hätte entfallen können). Man filtriert und wäscht den Niederschlag sorgfältig im Gooch, wobei man die Flüssigkeit und das Waschwasser auf etwa 100 c.cs.
erhitzt und zum Sieden bringt. 10 c.cs. gesättigte BaCl2-Lösung zugeben. 30 Minuten lang kochen lassen. Über Nacht stehen lassen. Durch einen Gooch filtern. Mit etwas E. HCl. und dann mit Wasser waschen. Trocknen, anzünden und wie üblich die BaSO4 abwiegen, die weiß und nicht mit Eisensalzen verunreinigt sein sollte.
Den Prozentsatz von S auf die übliche Weise berechnen. Da einige der verwendeten Reagenzien Schwefel enthalten können, muss eine Blindprobe mit den gleichen Mengen wie bei der eigentlichen Analyse durchgeführt werden, und der gefundene Schwefel muss vom vorherigen Ergebnis abgezogen werden.
ESTIMIERUNG VON SILIKON
Die hier angegebene Methode ist die von Drown und ist sowohl schnell als auch genau. Das Eisen wird in HNO3 gelöst, dann mit H2SO4 versetzt und bis zur Trockne eingedampft. Danach wird es in Lösung gebracht, wobei das Silizium als SiO2 im Rückstand verbleibt.
Details: – 2 g Bohrspäne werden abgewogen und in eine Platin- oder Porzellanschale überführt. Füge 30 c.cs. 8E. HNO3 Wenn die Wirkung scheinbar nachlässt, 20 c.cs. 18E. H2SO4 zugeben und eindampfen. (Blair empfiehlt einen sanften Warmluftstrom, der auf die Oberfläche der Flüssigkeit trifft. Die Luft wird erhitzt, indem man sie durch eine kleine Spirale aus einem über einem Bunsenofen erhitzten Kupferrohr leitet. Auf diese Weise wird die Verdampfung beschleunigt und ein Verspritzen verhindert.) Man setzt die Verdampfung fort, bis reichlich SO3-Dämpfe austreten. Abkühlen lassen und vorsichtig mit destilliertem Wasser auf 130 c.cs. verdünnen. Erhitzen, bis das gesamte FeSO4 aufgelöst ist. Filtrieren und zuerst mit etwas E. HCl und dann mit heißem Wasser waschen. Diese Filtration wird am besten mit einem aschefreien 7-cm-Filterpapier durchgeführt (die Asche wird durch Anzünden von zwei oder drei Papieren überprüft). Trocknen, in einen Platintiegel umfüllen, wie üblich anzünden und wiegen. In den Tiegel 5 ccm starke H2SO4 und 5 ccm starke HF geben. Vorsichtig bis zur Trockne eindampfen, wobei ein heißer Luftstrom die Verdampfung beschleunigt. Anzünden und erneut wiegen. Unter der Voraussetzung, dass H2SO4 und HF rein sind, entspricht die Gewichtsdifferenz SiO2. Die H2SO4 und die HF (insbesondere die HF) werden durch Verdampfen einer Blindprobe überprüft. Jeder gefundene Rückstand muss berücksichtigt werden.
Bestimmung von Phosphor
Auch hier werden von verschiedenen Stellen zahlreiche Methoden angegeben, von denen die meisten bei sorgfältiger Durchführung genaue Ergebnisse liefern. Die beiden für die technische Analyse am besten geeigneten Methoden sind die volumetrische Reduktionsmethode, die vom Unterkomitee (Herren Barba, Blair, Drown, Dudley und Shimer) des International Steel Standards Committee, U.S.A., ausgearbeitet wurde, und die modifizierte Reduktionsmethode, die von den Herren Dudley und Pease, Jour. Anal. Appl. Chem., vii. 108. Die erstere Methode wird in Blair’s Analysis of Iron ausführlich erörtert; die letztere Methode wird hier angegeben.
Das Eisen wird aufgelöst und das P als Phospho-Molybdat von Ammonium ausgefällt. Dieses wird gelöst und durch Einwirkung von Zn und H2SO4 wird das MoO3 reduziert; die reduzierte Flüssigkeit wird dann mit K2Mn2O8 (Standardlösung) titriert, und aus der Anzahl der verwendeten c.c. kann der P-Gehalt berechnet werden.
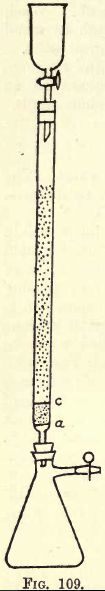
Details. -Wenn viel gearbeitet werden soll, ist ein Schüttelapparat erforderlich (siehe Kataloge für Chemikalien). Der Schüler kann jedoch das notwendige Schütteln auch von Hand durchführen. Bevor mit der Analyse begonnen wird, muss der Reduktionsapparat (eine Abwandlung des Jones-Reduktors) vorbereitet werden (siehe Abb. 109).
Auf a befindet sich eine fein gelochte Scheibe aus fester Platinfolie. Zwischen a und c befindet sich etwa ¾ Zoll sauberer weißer Sand, c ist eine weitere perforierte Platinscheibe.
Über dieser Scheibe wird das Rohr mit fein granuliertem amalgamiertem Zink gefüllt, das folgendermaßen hergestellt wird:-Löse 5 gg. Hg in 25 c.cs. starkem HNO3, verdünnt mit Wasser und füllt die Lösung auf 1 Liter auf. In diese Lösung gießt man ein halbes Kilo Zinkgranulat, das durch ein 20er, aber nicht durch ein 30er-Sieb passt. Eine oder zwei Minuten lang schütteln. Gieße die Lösung ab. Wasche und trockne das Zink, das nun amalgamiert ist. Trichter und Kolben werden wie gezeigt in die Apparatur eingesetzt.
Bereite die folgenden Reagenzien vor:-
(a) Die stark oxidierende Lösung von K2Mn2O8. 12 g. reines K2Mn2O8 in 1 Liter Wasser. Filtrieren und in Flaschen abfüllen.
(b) Die Molybdatlösung: Löse 50 g. MoO3 in 200 c.cs. NH4HO (S.G. .96). Man filtriert und fügt dem Filtrat 500 c.cs. HNO3 (S.G. 1,2). Vor Gebrauch mindestens 24 Stunden stehen lassen.
(c) Die saure Amnwniumsulfatlösung: Zu 500 c.c. destilliertem Wasser 27,5 c.cs. NH4HO (S.G. 0,96) und dann 24 c.cs. reines H2SO4 (S.G. 1,84) zu und verdünne auf 1000 c.cs.
(d) Die K2Mn2O8-Standardlösung: Löse 2 g kristallisiertes K2Mn2O8 in 1000 c.cs destilliertem Wasser. Die Lösung wird wie folgt standardisiert: Man wiegt 3 Partien von je 0,1 bis 0,3 g gründlich gereinigtem Eisendraht ab, dessen Eisengehalt bekannt ist. In einen 100-cm³-Erlenmeyerkolben umfüllen und je 40 ccm zugeben. 8E. H2SO4. Nach dem Auflösen 5 Minuten kochen, auf 150 ccm verdünnen, durch den Reduktor laufen lassen und waschen, wobei das Volumen auf 200 ccm gebracht wird, wie in der Analyse angegeben. Jede Partie wird mit K2Mn2O8 titriert. Die Ergebnisse sollten für metallisches Eisen auf 1/100 Milligramm genau übereinstimmen. Die Verunreinigungen im entnommenen Draht sind entsprechend zu berücksichtigen. Angenommen, 1 c.c. K2Mn2O8 = 0,0034923 g Fe, dann multipliziert man diesen Wert in Fe mit dem Verhältnis von MoO3 zu Fe, nämlich 0,9076, und das Produkt mit dem Verhältnis des vorhandenen P
zum MoO3, nämlich 0,019, so erhält man
1 c.c. K2Mn2O8 = 0,0000602 gm. P
Analyse
Wiegen Sie 1 gm. Bohrungen aus. In einen 200 c.c. Erlenmeyerkolben überführen. Füge 70 c.cs. 5E. HNO3. Wenn die Lösung vollständig ist, eine Minute kochen lassen und 10 ccm der „oxidierenden“ Lösung von K2Mn2O8 hinzufügen. Man kocht, bis die rosa Farbe verschwindet und sich MnO2 abscheidet. Man entfernt die Lösung und gibt unter Rühren nach und nach Kristalle von reinem (phosphorfreiem) FeSO4 hinzu, bis sich der Inhalt klärt. Die Lösung auf 80° C erhitzen (bei Vorhandensein von As auf 35° C). 75 c.c. Molybdatlösung bei einer Temperatur von 27° C zugeben. Den Kolben mit einem Gummistopfen verschließen und 5 Minuten lang schütteln. 5 Minuten lang stehen lassen. Dann filtriert man durch ein 9-cm-Filter und wäscht mit der sauren Ammonsulfatlösung, bis einige Tropfen des Waschwassers mit Ammoniumsulfid keine Farbe mehr ergeben.
Den Niederschlag auf dem Papier löst man mit 5 c.cs. NH4HO (S.G. 0,90) und 25 c.c. Wasser auf, wobei man die Lösung in den ursprünglichen Kolben zurücklaufen lässt und so den an den Rändern anhaftenden Niederschlag auflöst. Man wäscht, bis Filtrat und Waschmittel 150 c.c. ergeben. 10 c.cs. starke H2SO4 (S.G. 1.84) zugeben und auf 200 c.cs. verdünnen. Die Lösung ist nun zur Reduktion bereit.
100 c.cs. warme ~E/2 H2SO4 in den Trichter gießen. Schließe den Kolben an die Filterpumpe an und öffne die Klemme, so dass die Lösung fast, aber nicht ganz, aus dem Trichter fließt. Dann gibt man in den Trichter folgende Blindprobe – 5 c.cs. NH4HO (S.G. 0,90), 10 c.cs. H2SO4 (S.G. 1.84) und 50 c.cs. Wasser hinzu, die miteinander vermischt werden. Man öffnet wieder die Klammer, so dass diese Mischung fast aus dem Trichter läuft. Nun 200 c.cs. E/2 H2SO4 in den Trichter geben und fast durchlaufen lassen.
Den Kolben abnehmen, indem man zuerst den Absperrhahn des Trichters schließt. Man titriert den Inhalt des Kolbens mit K2Mn2O8. In der Regel werden dabei etwa 0,1 c.cs. Permanganat verbraucht, die bei künftigen Ablesungen abgezogen werden müssen.
Nun wird die zu reduzierende Lösung in den Trichter überführt. Befestige einen sauberen Kolben. Schließe die Filterpumpe an und starte sie. Öffne den Absperrhahn und die Klemme, so dass die Lösung fast durchläuft. Den Kolben, der die Lösung enthielt, spült man mit 100 c.cs. E/2 H2SO4 AUS. Diese wird in den Trichter gegeben und wie zuvor behandelt.
Schließlich werden weitere 100 c.cs. der Säure zugegeben und fast durchlaufen gelassen.
Die reduzierte Lösung im Filterkolben sollte nun hellgrün sein.
Entferne sie wie zuvor und titriere mit der Permanganatlösung. Das Grün verfärbt sich rosa-braun, dann rosa-gelb, dann farblos, und schließlich erhält man ein dauerhaftes Rosa (nach einer Minute Stehenlassen). Von dem erhaltenen Wert zieht man den Blindwert ab und berechnet den Prozentsatz des vorhandenen P aus den oben angegebenen Daten.
Anstelle dieser volumetrischen Methode ziehen es einige Chemiker vor, den gelben Phosphormolybdatniederschlag direkt zu wiegen. Für Einzelheiten siehe Blair’s Analysis of Iron, S. 108.
Anmerkung: Der Student sollte, wo immer möglich, die Vorteile von Verweisen auf spezielle Behörden nutzen. Zu diesem Zeitpunkt sollte er in der Lage sein, solche Materialien zu konsultieren, zu vergleichen und bis zu einem gewissen Grad vernünftig zu nutzen. Kein einziges Lehrbuch kann eine umfassende Behandlung des Themas „Eisen und Stähle“ oder auch nur eines der in diesem Abschnitt behandelten Themen bieten; daher müssen die angegebenen Referenzen zusammen mit der aktuellen Literatur von dem Analytiker, der in der technischen Arbeit brillieren will, sorgfältig gelesen werden. Die kolorimetrische Bestimmung des gebundenen Kohlenstoffs nach der Eggertz-Methode wurde angegeben; Mangan kann in ähnlicher Weise nach der kolorimetrischen Methode von Peter oder nach der Acetat-Methode (siehe Blair usw.) bestimmt werden.