I detta kapitel presenteras sådant material som kan ge studenten en inblick i de analysmetoder som används vid teknisk undersökning av järn och stål. Arbetets karaktär förhindrar en detaljerad behandling av ämnet – en behandling som kan kräva en uppskattning av ett eller flera av följande ämnen: Kol (fritt och kombinerat), svavel, kisel, fosfor, mangan, titan, koppar, nickel, kobolt, krom, aluminium, arsenik, antimon, tenn, volfram, vanadin, kväve, järn. I allmänhet är de uppskattningar som krävs mest av kol, svavel, kisel och fosfor. Av de övriga grundämnen och föreningar som nämns krävs bestämningar av ett eller flera för specialstål. För information om dessa bestämningar hänvisas studenten till Chemical Analysis and Foundry Chemistry, av Crobaugh; The. Chemical Analysis of Iron, av Blair ; ”Carbon in Steel by Direct Combustion”, av Blount, i The Analyst, januari 1902; ”Sulphur in Wrought Iron and Steel”, av Auchy, i Jour. Amer. Chem. Soc., mars 1901, och andra artiklar i samma tidskrifter. Den studerande som vill gå vidare bör om möjligt få tillgång till Campbell, Drowns och andras uppsatser och artiklar som publiceras från tid till annan i de olika kemiska och metallurgiska tidskrifterna.
Eftersom studentens tid är begränsad kan han för närvarande skjuta upp uppskattningen av kisel och fosfor, även om dessa ges på grund av deras betydelse både för metallurgen och gjuteristen.
För att studenten skall få ett mer grundligt grepp om ämnet är det inte onödigt att göra några anteckningar om sammansättningen och egenskaperna hos de ämnen som behandlas. När det gäller de olika elementens inverkan på stålet, se The Manufacture and Properties of Structural Steel, av H. H. Campbell.
Kol finns i järn i tre tillstånd – grafitiskt, upplöst och kombinerat. Förutom dessa har andra former identifierats med hjälp av mikroskopet.
Svavel finns i järn huvudsakligen som sulfiden FeS. som är löslig i smält järn.
Fosfor finns som järnfosfid, som är helt löslig i det smälta järnet.
Silikon bildar silicid av järn, som också är lösligt i smält järn.
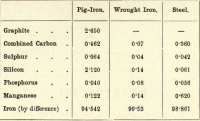
Av dessa fyra grundämnen är kol alltså det enda som kan existera i fritt tillstånd. Variationerna i proportionerna av de olika förekommande grundämnena är nästan oändliga, men följande korta tabell ger den ungefärliga sammansättningen av tackjärn, smidesjärn och stål, även om var och en av dessa är föremål för betydande variationer.
Den studerande ska uppskatta följande:
(1) Kol,
(a) Totalt.
(b) Grafitiskt.
(c) Kombinerat.
(2) Svavel.
(3) Kisel. (Om tiden tillåter.)
(4) Fosfor. (Om tiden tillåter.)
KOL TOTALT
I denna beräkning omvandlas kolet till koldioxid som absorberas i kaustisk kaliumklorid. Från vikten av den CO2 som på detta sätt erhålls beräknas kolet.
Vid första anblicken verkar det enklaste förfarandet vara att antända järn- eller stålborrningarna direkt i en ström av syre och absorbera den CO2 som på detta sätt bildas i KHO. Tyvärr har denna metod hittills antingen visat sig vara felaktig, eller när fullständig förbränning har erhållits har den apparatur som krävs för att motstå den höga temperaturen eller andra variationer i behandlingen inte lämpat sig för tekniskt arbete (se artiklar av Blount i The Analyst). Den studerande kommer att finna att den här givna metoden inte alls är idealisk ur teknisk synvinkel när det gäller bekvämlighet och snabbhet, och det verkar troligt att den inom en nära framtid kommer att ersättas av någon snabbare direkt oxidationsmetod.
Metod som antagits.- Vid hänvisning till de många arbetena i detta ämne kommer man att finna en stor variation av metoder. Den metod som anges här kommer med normal noggrannhet att ge korrekta resultat. Kortfattat är den följande:-
Järnet löses upp i en lösning av dubbelklorid av kalium och koppar, som görs sur med HCl. Metallisk koppar fälls ut och löses på nytt; järnet löses upp och kolet lämnas i suspension. Det samlas sedan upp och antänds i förbränningsugnen med syre och den koldioxid som utvecklats vägs.
Lösning av järnet.-Väga upp 1 gram råjärnborrning. Överför till en 300 c.c. bägare. Tillsätt 100 c.cs. CuCl2,2KCl,2H2O-lösning, som framställs på följande sätt. Lös 149,1 delar KCl och 170,3 delar kristalliserat CuCl2, 2H2O i vatten. Avdunsta och kristallisera ut dubbelkloriden. Lös 300 g av dubbelsaltet i destillerat vatten. Filtrera genom antänd asbest och förvara det i glasflaskor.
Till innehållet i bägaren tillsätt 7 c.cs. HCl för att göra lösningen sur. Rör om med jämna mellanrum tills järnet är löst. Placera bägaren och dess innehåll mot slutet av lösningen på ett vattenbad vid en temperatur av ca 60° C. Följande reaktioner äger rum-Fe + CuCl2 = FeCl2 + Cu och Cu + CuCl2 = 2CuCl. KCl underlättar helt enkelt lösningen av den utfällda kopparen. Inom cirka 40 minuter efter tillsättningen av de dubbla kloriderna bör lösningen vara nästan fullständig och det mesta av kopparn lösts upp. Skölj ner bägarens sidor med lite sura dubbelklorider. Till lösningen tillsätt lite antänd asbest för att sedimentera det kolhaltiga materialet och förhindra att det täpper till filtret (enligt Barbas rekommendation).
För filtrering är speciella platinabåtar, utrustade enligt Goochs degelprincip, mycket praktiska. Eleven kan dock filtrera bort de kolhaltiga ämnena med hjälp av en Goochdegel med hjälp av artificiell sugning, där de kolhaltiga ämnena sköljs in med en vattenstråle efter det att vätskan har passerat genom filtret. Tvätta försiktigt kolet på filtret med varmt vatten. Torka degeln och
innehållet i luftugn vid 100 C. Det kolhaltiga materialet är nu redo för antändning.
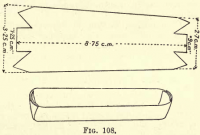
Oxidation av kolet.-Förbered en platinabåt genom att skära till en bit platinafolie enligt fig. 108, vik upp sidorna och ändarna så att de bildar ett tråg. Överför kolhaltiga ämnen och asbest från Gooch till båten.
Förbränningsugnen, tillbehör och armaturer måste ställas i ordning. Syrgasreningsapparaten används återigen, men den här gången är den försedd med ett trevägsrör, med kranar insatta mellan lagret och renarna. Detta gör det möjligt att dra en luftström genom apparaten. Förbränningsröret kan vara av hårt Jena-glas, porslin eller platina. Två U-rör används mellan ugnen och kaliumlamporna. Den del av det första röret som ligger närmast ugnen innehåller vattenfritt CuSO4 och den andra delen vattenfritt CuCl. Det andra U-röret innehåller torkat CaCl2. Dessa två rör utgör ”reningståget”. CuCl absorberar all Cl och de andra ämnena all H2O. Denna uppsättning kan användas för många bestämningar. Kaliumlamporna och skyddsrören följer efter, och en aspirator bör finnas till hands för att dra en luftström genom apparaten när det behövs. Kaliumlamporna laddas med 8E. KHO och skyddsröret med CaCl2. Testa ugnen och glödlamporna som tidigare beskrivits (se Kol och koks), röret laddas som i skissen, båten hålls för närvarande i luftugnen vid 100° C.
När allt är klart, brännarna har varit släckta en tid, sätts båten och innehållet in. Brännarna tänds från den främre änden, arbetar successivt bakåt, och en långsam ström av syre, ungefär två bubblor per sekund, har tidigare slagits på tills röret är fullt av syre. Reglera temperaturen tills båten är matt röd, och om lösningen i glödlamporna visar tecken på att rinna tillbaka till ugnen, öka strömmen av syre till tre eller fyra bubblor per sekund.
Från det att båten sätts in räcker det med cirka femtio minuter för en fullständig förbränning. Stäng av syret och låt en luftström passera i tio minuter.
Potaskulorna och skyddsröret tas nu bort och vägs, och kolet beräknas som vanligt.
(b) Grafitiskt kol – Järnet löses av vissa i HCl, av andra i HNO3 när det grafitiska kolet blir kvar som en rest. För tackjärn ger båda metoderna, med försiktighet, goda resultat, men för stål som innehåller grafit rekommenderar Blair lösning i salpetersyra. (För denna metod rådfråga Blair.)
Väga ut 5 gram borrhål av tackjärn. Lös upp i 50 c.cs. SE. HCl med hjälp av mild värme. Koka i några minuter. Späd till 100 c.cs. (nästan). Filtrera genom en Goochdegel. Tvätta väl med varmt vatten och sedan med kokande E. KHO. (Detta löser upp eventuellt SiO2.) Tvätta igen med varmt vatten för att avlägsna KHO. Torka degeln och dess innehåll.
Skatta kolet som tidigare genom förbränning och beräkna procentandelen som vanligt.
(c) Kombinerat kol (genom skillnad).-Det totala kolet och det grafitiska kolet är kända, och det kombinerade kolet erhålls genom att subtrahera det grafitiska från det totala kolet.
För direkta metoder för uppskattning konsultera de nämnda auktoriteterna.
ESTIMERING AV SVAVEL I JÄRN & STÅL
Det råder avsevärda meningsskiljaktigheter om den bästa metoden för uppskattning av svavel i järn och stål. Den gamla metoden med vattenlösning och BaCl2-utfällning erkänns som mycket felaktig, men långsam lösning i HNO3, med mycket lite eller ingen HCl närvarande, följt av noggrann utfällning med BaCl2 i närvaro av ett klart överskott av HCl och med vederbörlig omsorg när det gäller tid och förhållanden för utfällningen och försiktighetsåtgärder mot att utfällningen kontamineras av järn – med dessa och omsorg kan man få goda resultat. Blair, å andra sidan, rekommenderar lösning i HCl, där S utvecklas som H2S, som absorberas i en (alkalisk) lösning av Pb(NO3)2 och bildar PbS, som löses upp i HCl + KClO3, och S fälls ut som BaSO4. För ytterligare metoder se Blair, Stillman, Auchy, Crobaugh och Drown. En annan vanligt förekommande metod är att S utvecklas som H2S, följt av absorption i kadmiumkloridlösning. Den utfällda kadmiumsulfiden löses upp i HCl och S uppskattas genom titrering med en jodlösning, eller ännu vanligare, H2S absorberas i Br. vatten och fälls sedan ut som BaSO4 eller absorberas i NaOH och titreras med jod; den sistnämnda är den vanligaste metoden. (Se Blair.) Följande metod ges här:-
Oxidation med HNO3 (den s.k. Aqua Regia-metoden).-Väga upp 5 g borrningar och för över dem till en djup 200 c.c. bägare. Tillsätt försiktigt ca 40 c.cs. 16E. HNO3, i omgångar om ca 10 c.c. i taget, genom att täcka bägaren med ett stort klockglas och se till att det inte sker för våldsamt. När det verkar som om det upphör, kontrollera om alla partiklar har lösts upp (utom kol). Om inte, värm på sandbadet och tillsätt 3 eller 4 droppar 16E. HCl, och värm tills de är lösta.
När lösningen är fullständig, tillsätt lite Na2CO3 för att omvandla eventuellt H2SO4 till Na2SO4, som inte är flyktigt vid avdunstning.
Ta bort sandbadet och tillsätt 5 c.c. stark HCl utöver det som behövs för att precis lösa upp eventuella järnföreningar som fällts ut av Na2CO3. Filtrera bort SiO2 och C. Tvätta väl. Indunsta till torrhet för att göra SiO2 olösligt. Ta upp med HCl och avdunsta tills Fe2Cl6 börjar kristallisera ut. Tillsätt sedan 5 c.cs. HCl. och filtrera om det finns någon rest. (Om det inte finns någon rest finns det inget SiO2 i lösningen, och indunstningen kunde ha utelämnats). Filtrera och tvätta försiktigt utfällningen i Gooch, för upp vätskan och tvätten till ca 100 c.cs.
Hetta upp till kokning. Tillsätt 10 c.c. mättad lösning av BaCl2. Koka i 30 minuter. Låt stå över natten. Filtrera genom en Gooch. Tvätta med lite E. HCl. och sedan med vatten. Torka, antänd och väg som vanligt BaSO4, som ska vara vitt och inte förorenat med järnsalter.
Beräkna den procentuella andelen S på vanligt sätt. Eftersom en del av de använda reagenserna kan innehålla svavel måste en blankprovtagning göras med samma mängder som vid den egentliga analysen, och eventuellt påträffat svavel dras av från det tidigare resultatet.
ESTIMERING AV SILIKON
Metoden som ges här är Drowns metod, och är både snabb och exakt. Järnet löses upp i HNO3, följt av H2SO4, med avdunstning till torrhet. Detta följs av lösning, varvid silikonet kvarstår i restprodukten som SiO2.
Detaljer: – Väg upp 2 g borrningar och överför dem till en platina- eller porslinsskål. Tillsätt 30 c.cs. 8E. HNO3 När verkan till synes upphör, tillsätt 20 c.cs. 18E. H2SO4 och avdunsta. (Blair rekommenderar att en lätt blåst av varm luft spelar på vätskans yta. Luften värms upp genom att den leds genom en liten spiral av kopparrör som värms upp över en bunsen. Avdunstningen påskyndas på så sätt och spiralbildning förhindras). Fortsätt avdunstningen tills rikliga ångor av SO3 avges. Låt svalna och späd försiktigt med destillerat vatten till 130 c.cs. Värm tills allt FeSO4 är upplöst. Filtrera och tvätta först med lite E. HCl och sedan med varmt vatten. Denna filtrering utförs bäst med ett 7 cm. askfritt filterpapper (kontrollera askan genom att antända två eller tre av pappren). Torka, överför till en degel av platina, antänds som vanligt och vägs. Till degeln tillsätt 5 c.c. starkt H2SO4 och 5 c.c. starkt HF. Avdunsta försiktigt till torrhet, med hjälp av en varm luftstöt för att påskynda avdunstningen. Tänd och väga på nytt. Om H2SO4 och HF är rena, representerar viktskillnaden SiO2. Kontrollera H2SO4 och HF (särskilt det sistnämnda) genom att förånga ett blankprov. Eventuella rester måste beaktas.
ESTIMATION AV FOSFOR
Även här ges många metoder av olika auktoriteter, och de flesta av dem ger korrekta resultat om de följs noggrant. De två metoder som lämpar sig bäst för teknisk analys är den volymetriska reduktionsmetoden som utarbetats av underkommittén (Barba, Blair, Drown, Dudley och Shimer) i International Steel Standards Committee, U.S.A., och den modifierade reduktionsmetoden som ges av Dudley och Pease, Jour. Anal. Appl. Chem., vii. 108. Den förstnämnda metoden diskuteras utförligt i Blair’s Analysis of Iron; den sistnämnda metoden ges här.
Järnet löses upp och P fälls ut som fosfomolybdat av ammonium. Detta löses upp, och genom inverkan av Zn och H2SO4 reduceras MoO3, och den reducerade vätskan titreras sedan med K2Mn2O8 (standardlösning), och från det antal c.c. som används kan P-halterna beräknas.
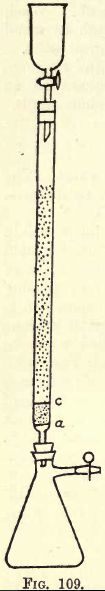
Detaljer. -Om mycket arbete ska utföras är en skakapparat nödvändig (se kataloger över kemiska förnödenheter). Eleven kan dock utföra den nödvändiga skakningen för hand. Innan analysen påbörjas måste reduktionsapparaten (en modifiering av Jones reductor) förberedas (se fig. 109).
I a finns en finperforerad skiva av kraftig platinafolie. Mellan a och c finns ungefär ¾ tum ren vit sand, c är en annan perforerad platinaskiva.
Ovanför denna skiva fylls röret med fint granulerad amalgamerad zink, som framställs på följande sätt: – Lös upp 5 gram. Hg i 25 c.c. starkt HNO3, späd med vatten och fyll på lösningen till 1 liter. I denna lösning
hälls ett halvt kilo granulerad zink som passerar en 20 men inte en 30 sikt. Skaka i en eller två minuter. Häll av lösningen. Tvätta och torka zinken, som nu är amalgamerad. Tratten och kolven monteras på apparaten enligt bilden.
Förbered följande reagenser :-
(a) Den starkt oxiderande lösningen av K2Mn2O8. 12 g ren K2Mn2O8 i 1 liter vatten. Filtrera och fyll på flaska.
(b) Molybdatlösning: Lös upp 50 gms. MoO3 i 200 c.c. NH4HO (S.G. .96). Filtrera och tillsätt till filtratet 500 c.cs. HNO3 (S.G. 1,2). Låt stå minst 24 timmar före användning.
(c) Den sura amnwniumsulfatlösningen: Till 500 c.c. destillerat vatten tillsätts 27,5 c.c. NH4HO (S.G. 0,96) och sedan 24 c.c. rent H2SO4 (S.G. 1,84) och späd till 1000 c.c.
(d) Standard K2Mn2O8-lösning: Lös upp 2 g kristalliserat K2Mn2O8 i 1000 c.c. destillerat vatten. Standardisera lösningen på följande sätt: Väg ut 3 partier på 0,1 till 0,3 g vardera av noggrant rengjord järntråd, vars järninnehåll är känt. Överför till 100 c.c. Erlenmeyerkolv och tillsätt till varje kolv 40 c.c. 8E. H2SO4. Koka upp i 5 minuter när de är lösta, späd till 150 ccm, passera genom reduktorn och tvätta, för att få upp volymen till 200 ccm, i enlighet med anvisningarna i analysen. Titrera varje parti med K2Mn2O8. Resultaten bör överensstämma för metalliskt järn med en noggrannhet på 1/100 milligram. Gör den erforderliga avvägningen för föroreningar i den tagna tråden. Anta att 1 c.c. K2Mn2O8 = 0,0034923 gm Fe, multiplicera sedan detta Fe-värde med förhållandet mellan MoO3 och Fe, nämligen 0,9076, och produkten med förhållandet mellan P
närvarande och MoO3, nämligen 0,019, så får vi
1 c.c. K2Mn2O8 = 0,0000602 gm. P
Analys
Väga ut 1 gm borrningar. Överför till en 200 c.c. Erlenmeyerkolv. Tillsätt 70 c.cs. 5E. HNO3. När lösningen är fullständig, koka en minut och tillsätt 10 c.c. av den ”oxiderande” lösningen av K2Mn2O8. Koka tills den rosa färgen försvinner och MnO2 avskiljs. Ta bort lösningen och tillsätt gradvis under omrörning kristaller av rent (fosforfritt) FeSO4 tills innehållet klarnar. Värm lösningen till 80° C (om As är närvarande, till 35° C). Tillsätt 75 c.c. av molybdatlösningen vid en temperatur på 27° C. Stäng kolven med en gummipropp och skaka i 5 minuter. Låt stå i 5 minuter. Filtrera sedan genom ett 9 cm, filter och tvätta med den sura amm. sulfatlösningen tills några droppar av tvätten inte ger någon färg med ammoniumsulfid.
Upplös utfällningen på pappret med 5 c.cs. NH4HO (S.G. 0,90) och 25 c.c. vatten och låt lösningen rinna tillbaka till den ursprungliga kolven, vilket löser upp alla utfällningar som fastnar på sidorna. Tvätta tills filtrat och tvättvatten uppgår till 150 c.cs. Tillsätt 10 c.c. starkt H2SO4 (S.G. 1,84) och späd till 200 c.c. Lösningen är nu klar för reduktion.
Häll 100 c.c. varmt ~E/2 H2SO4 i tratten. Anslut kolven till filterpumpen och öppna klämman så att lösningen nästan, men inte helt, rinner ut ur tratten. Tillsätt sedan följande blank-5 c.cs. NH4HO (S.G. 0,90), 10 c.cs. H2SO4 (S.G. 1,84) och 50 c.c. vatten, som blandas. Öppna återigen klämman så att denna blandning nästan rinner ut ur tratten. Tillsätt nu 200 c.cs, E/2 H2SO4 till tratten, och låt den nästan rinna igenom.
Deskappa kolven, stäng först stoppkranen olja tratten. Titrera kolvens innehåll med K2Mn2O8. I allmänhet förbrukas på detta sätt ca 0,1 c.c. permanganat, och denna mängd måste dras av från framtida avläsningar.
Transferera nu den lösning som skall reduceras till tratten. Fäst en ren kolv. Anslut och starta filterpumpen. Öppna avstängningskranen och klämman så att lösningen nästan rinner igenom. Tvätta ur kolven som innehöll lösningen med 100 c.cs. E/2 H2SO4. Tillsätt detta till tratten och behandla som tidigare.
Till sist, tillsätt och kör nästan igenom ytterligare 100 c.c. av syran.
Den reducerade lösningen i filterkolven bör nu vara ljusgrön.
Ta bort den som tidigare och titrera med permanganatlösningen. Det gröna förändras till rosabrunt, sedan rosagult, sedan färglöst och slutligen erhålls en permanent rosa färg (efter att ha stått en minut). Från den erhållna avläsningen drar man av blankavläsningen och beräknar den procentuella andelen P som finns med hjälp av ovanstående data.
Istället för denna volymetriska metod föredrar vissa kemister att väga den gula fosfomolybdatutfällningen direkt. För detaljer se Blair’s Analysis of Iron, s. 108.
Note.-Studenten bör, närhelst det är möjligt, dra nytta av hänvisningar till särskilda auktoriteter. Vid denna tidpunkt bör han vara kapabel att konsultera, jämföra och i viss mån använda sådant material på ett klokt sätt. Ingen lärobok kan på något sätt ge en heltäckande behandling av järn och stål, eller för den delen av något av de ämnen som behandlas i detta avsnitt; därför måste sådana referenser som ges, tillsammans med aktuell litteratur, noggrant läsas av den analytiker som vill utmärka sig i tekniskt arbete. Den kolorimetriska bestämningen av kombinerat kol med Eggertz’ metod har angetts ; mangan kan bestämmas på liknande sätt med Peters kolorimetriska metod, eller med acetatmetoden (se Blair, etc.).