W tym poście przedstawiamy karbaminiany jako użyteczną grupę ochronną dla amin, szczególnie w kontekście tworzenia peptydów.
Oto szybki obraz podsumowujący to, do czego przejdziemy w tym poście, z większą ilością szczegółów poniżej.
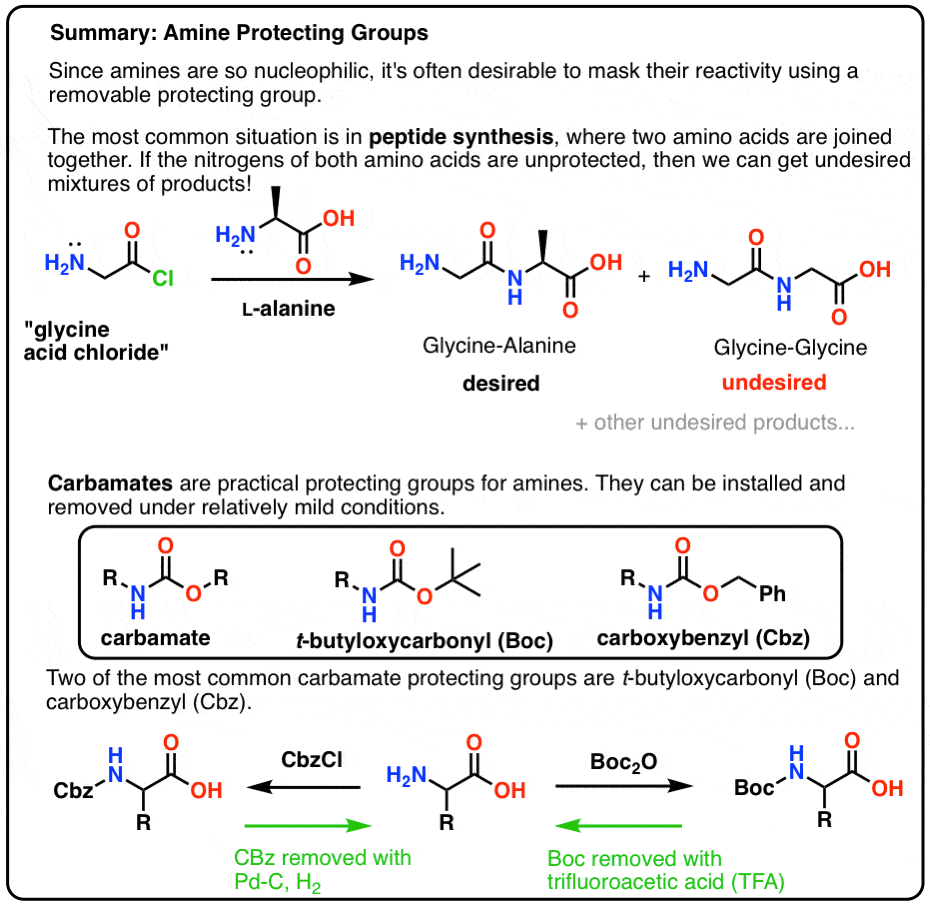
Table of Contents
- An Introduction To Simple Peptide Synthesis
- Let’s Mix Up A Batch of Gly-Ala
- A Very Bad Initial Plan
- „Hark! What Crap Is This?”
- A Protective Hat For NH2
- Mate! Użyj karbaminianu, kolego!
- Boc and CBz Are The Bees Knees
- Installation and Removal of the „Boc” Protecting Group
- Installation and Removal of The CBz (or „Z”) Carbamate Protecting Group
- A Simple Peptide Synthesis Using Carbamate Protecting Groups
- It Keeps Going…
- Notes
- (Advanced) References and Further Reading
An Introduction To Simple Peptide Synthesis
W ostatnim poście przeszliśmy przez 3 popularne sposoby robienia amidów:
- Dodawanie amin do halogenków acylowych / bezwodników
- Częściowa hydroliza nitryli
- Sprzęganie kwasów karboksylowych z aminami przy użyciu środka odwadniającego, takiego jak DCC (N,N’-dicykloheksylokarbodiimid).
W tym poście brakowało jakiejkolwiek wzmianki o syntezie pradziadka najbardziej użytecznych połączeń amidowych znanych ludzkości, a przez to rozumiem peptydy.
Wiązanie peptydowe to nazwa, którą nadajemy wiązaniu amidowemu łączącemu dwa aminokwasy.
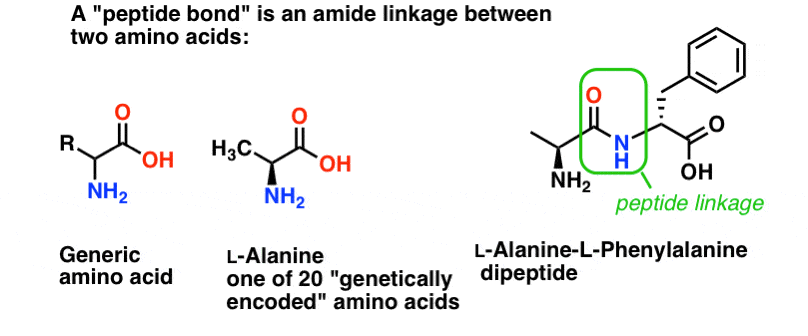
A aminokwasy są ważne, ponieważ… czekaj. Już wiesz dlaczego aminokwasy są ważne, prawda? Jeśli teraz tego nie wiesz, wróć po przeczytaniu tego.
Let’s Mix Up A Batch of Gly-Ala
Postarajmy się zastosować niektóre z naszych nowo znalezionych umiejętności syntezy amidów, aby spróbować zbudować naprawdę prosty dipeptyd, glicynę-alaninę. Jeśli uda nam się teraz zbudować prosty dipeptyd, będziemy mogli wykorzystać to, czego się nauczyliśmy, aby pokazać, jak powstają jeszcze bardziej złożone peptydy w późniejszym poście. Czy wiesz, że laureat Nagrody Nobla Bruce Merrifield zsyntetyzował insulinę, łącząc po jednym aminokwasie na raz? Tak, naprawdę.
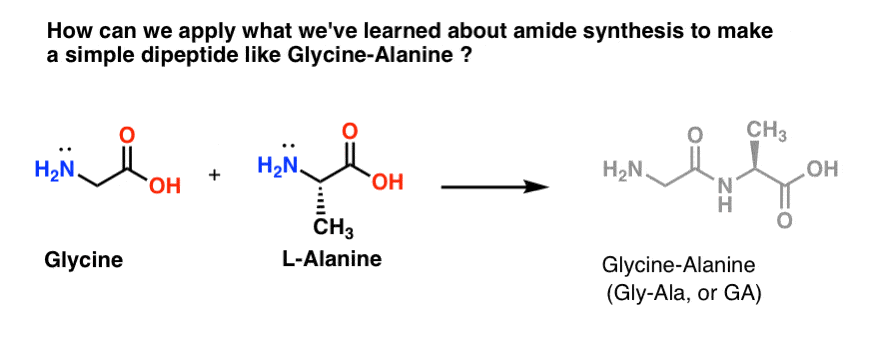
Z trzech metod, które wymieniliśmy do robienia amidów, tylko dwie są potencjalnie przydatne do tworzenia wiązań peptydowych: 1) metoda chlorku kwasu, lub 2) synteza poprzez czynnik sprzęgający, taki jak DCC. .
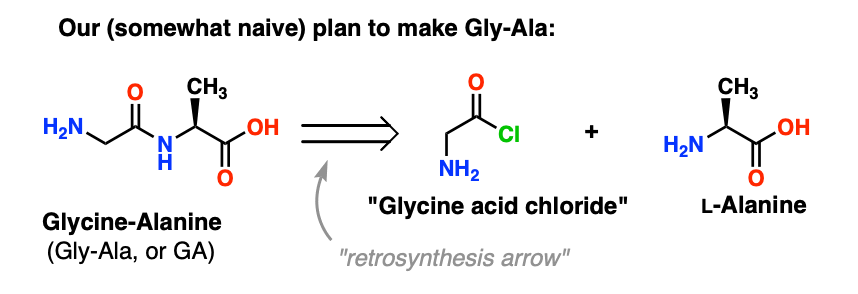
3. Bardzo zły plan początkowy
Nasze pierwsze podejście do syntezy peptydów będzie wiązało się z pędzeniem naprzód z bardzo naiwnym planem ataku i nadzieją, że w końcu wszystko się uda. Trzymajcie się ludzie, bo zrobi się bałagan.
Zaczniemy od zaproponowania metody chlorku kwasu do syntezy Gly-Ala.
Patrząc wstecz od dipeptydu Gly-Ala, nasz plan przewiduje syntezę wiązania peptydowego poprzez reakcję alaniny z chlorkiem kwasu pochodzącego z glicyny:
Z tym planem jest mały problem. Może być trochę trudny do zauważenia na początku.
Załóżmy, że zrobiliśmy chlorek kwasu z glicyny* , i mamy „chlorek kwasu z glicyny” i alaninę razem w tej samej kolbie, wraz z pewnym nadmiarem zasady, aby przyspieszyć sprawy.
Narysujemy reakcję w kierunku do przodu:
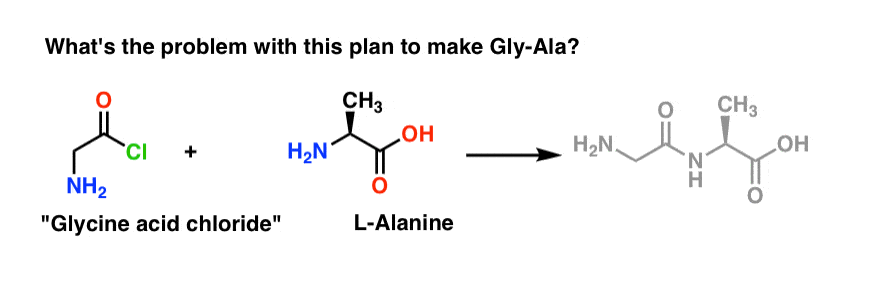
Co mogłoby pójść nie tak?
„Hark! What Crap Is This? „
Nasz plan jest zrobić roztwór chlorku kwasu glicyny (1 równoważnik molowy), a następnie cierpliwie czekać w kolbie, aż dodamy równoważnik molowy alaniny, gdzieupon będzie reagować z nukleofilowej grupy NH2 alaniny.
Problem z naszym planem polega na tym, że nie mamy do czynienia z pojedynczą cząsteczką „chlorku kwasu glicynowego” – mamy do czynienia z czymś około mola (6.02 x 1023 cząsteczek) tego kwasu. A chlorek kwasu glicynowego ma już nukleofilową grupę NH2!
„Chlorek kwasu glicynowego” tak narysowany, nie jest stabilną cząsteczką, bo może reagować sam ze sobą.
To oznacza, że roztwór chlorku kwasu glicynowego pozostawiony samemu sobie utworzy polimer glicyny o strukturze Gly-Gly-Gly-Gly…
Nawet roztwór chlorku kwasu glicynowego w obecności alaniny nie tylko utworzy pożądane Gly-Ala, ale również Gly-Gly (z dołączonym halogenkiem acylowym), który może dalej czynić więcej szkód z innym nukleofilem, czy to Gly czy Ala:
(i nie, nie ma wiele, aby odróżnić NH2 „chlorku kwasu glicynowego” od NH2 alaniny. Są mniej więcej tak samo reaktywne.)
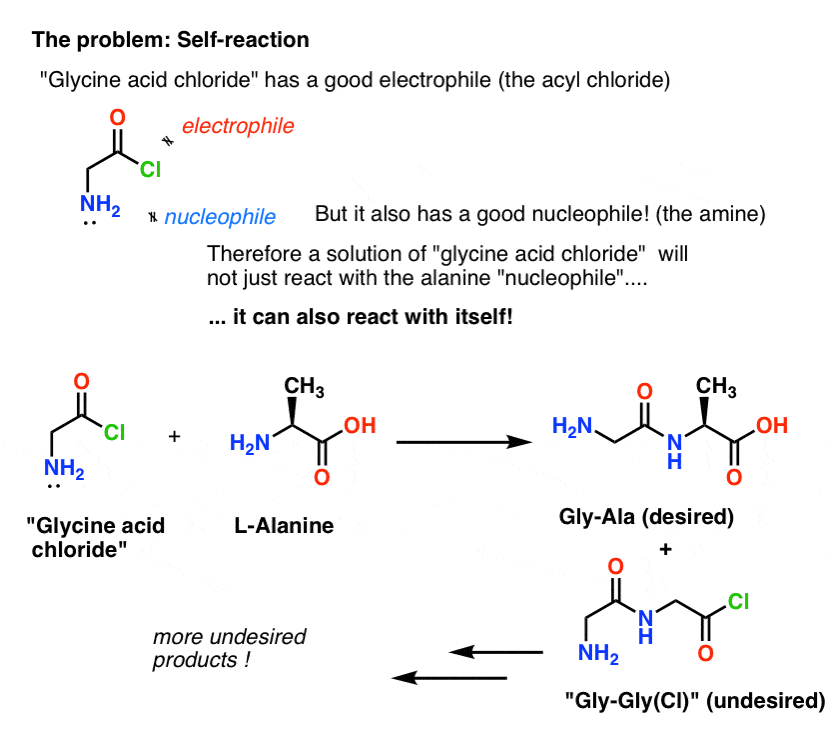
Nauka tutaj jest taka, że kiedy masz roztwór cząsteczki zawierającej zarówno nukleofil jak i elektrofil, może ona samoczynnie reagować. Istnieje nazwa dla tego procesu, która może brzmieć znajomo: polimeryzacja.
Jak więc temu zapobiec?
Kapelusz ochronny dla NH2
Najlepszym sposobem jest „przykrycie” azotu w jakiś sposób grupą ochronną (PG), która sprawia, że grupa NH2 nie jest nukleofilowa. Powinna ona również posiadać następujące właściwości:
- łatwo i selektywnie instalowana
- nieodporna na pożądane warunki reakcji (np. SOCl2 do wytworzenia chlorku kwasu z kwasu karboksylowego)
- łatwo i selektywnie usuwane bez wpływu na produkt końcowy
Wcześniej widzieliśmy strategie grup ochronnych, szczególnie w przypadku alkoholi i stosowania odczynników Grignarda.
Oto jak może wyglądać strategia grup ochronnych dla naszej syntezy „Gly-Gly”. Montujemy grupę ochronną („PG”) na glicynie, a następnie tworzymy chlorek kwasu. PG powinna być tak dobrana, aby azot nie był nukleofilowy (tzn. nie będzie reagował z chlorkiem kwasu).
Możemy wtedy utworzyć nasze wiązanie peptydowe z niechronioną alaniną, a następnie usunąć PG w łagodnych warunkach.
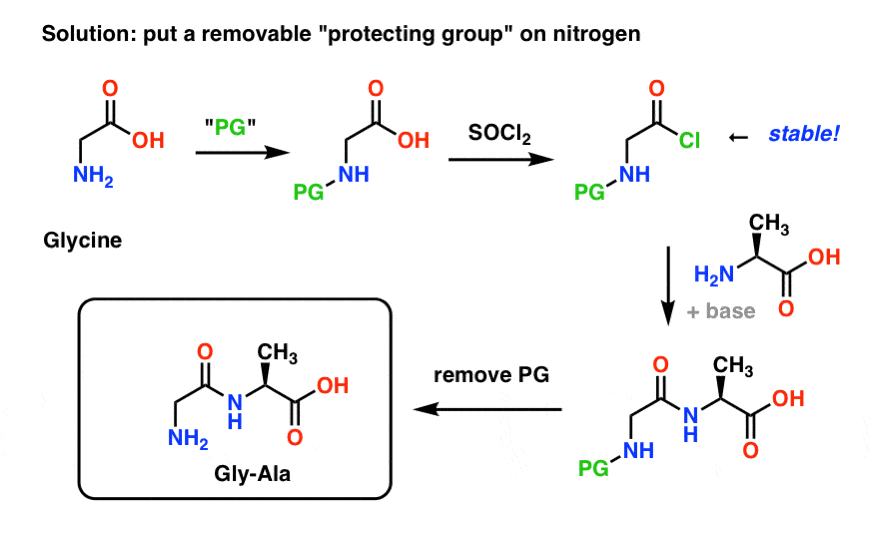
Jedną ze strategii grup ochronnych dla azotu, którą już zbadaliśmy, jest synteza Gabriela, która wykorzystuje ftalimid (można myśleć o ftalimidzie jako chronionym azocie). To faktycznie zostało wykorzystane do syntezy Gly-Gly („glycylglycine”)! Jednym z problemów jest jednak to, że stosunkowo surowe warunki (dużo ciepła) są wymagane, aby zarówno zainstalować i usunąć grupę ftalimidu, a to nie jest bardzo zdrowe środowisko dla przetrwania wrażliwych, chiralnych aminokwasów, które mogą łatwo racemize.
Innym potencjalnym wyborem jest ochrona azotu jako amid, ale amidy rozszczepienia może wymagać surowe warunki zbyt. Ponadto, ponieważ i tak próbujemy tu wykuć wiązanie amidowe (peptyd), możemy mieć problemy z selektywnością jego usunięcia – niszcząc wieś, aby ją uratować.
Mate! Użyj karbaminianu, kolego!
Najpopularniejszym wyborem grupy ochronnej dla azotu aminowego jest grupa funkcyjna karbaminianu. Karbaminian wygląda jak dziecko bękarta estru i amidu, z N i O flankującymi karbonyl.
Atrogen karbaminianu jest stosunkowo mało nukleofilowy, a ponadto, karbaminiany są:
- łatwo montowane na azocie
- obojętne na wiele różnych warunków reakcji
- łatwo usuwane bez wpływu na istniejące grupy amidowe
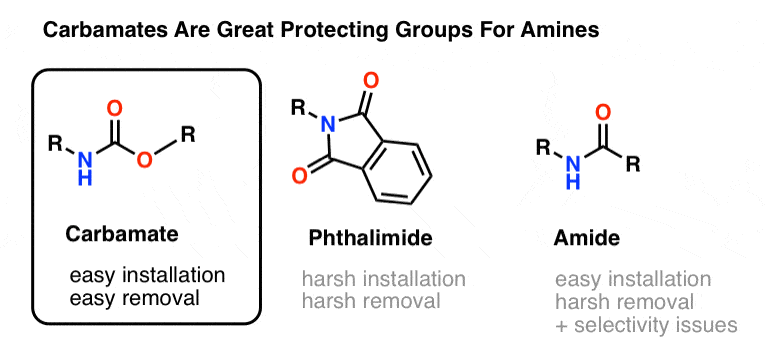
Dzięki temu doskonale nadają się do naszych celów.
Boc and CBz Are The Bees Knees
Dwie popularne karbaminianowe grupy ochronne to Boc (t-Butyloxycarbonyl) i CBz (carboxybenzyl).
Dla naszych celów, te dwie grupy ochronne mogą być uważane za mniej więcej równoważne, ponieważ każda z nich może być skutecznie wykorzystana do syntezy peptydów.
Kluczowa różnica polega na tym, jak są one usuwane (tj. etap „deprotekcji”). Wybór pomiędzy jednym a drugim staje się kluczowy, gdy masz złożoną cząsteczkę z wieloma grupami ochronnymi; to podpada pod kategorię „zaawansowanej strategii syntetycznej”, która jest bardziej tematem dla Org 3.
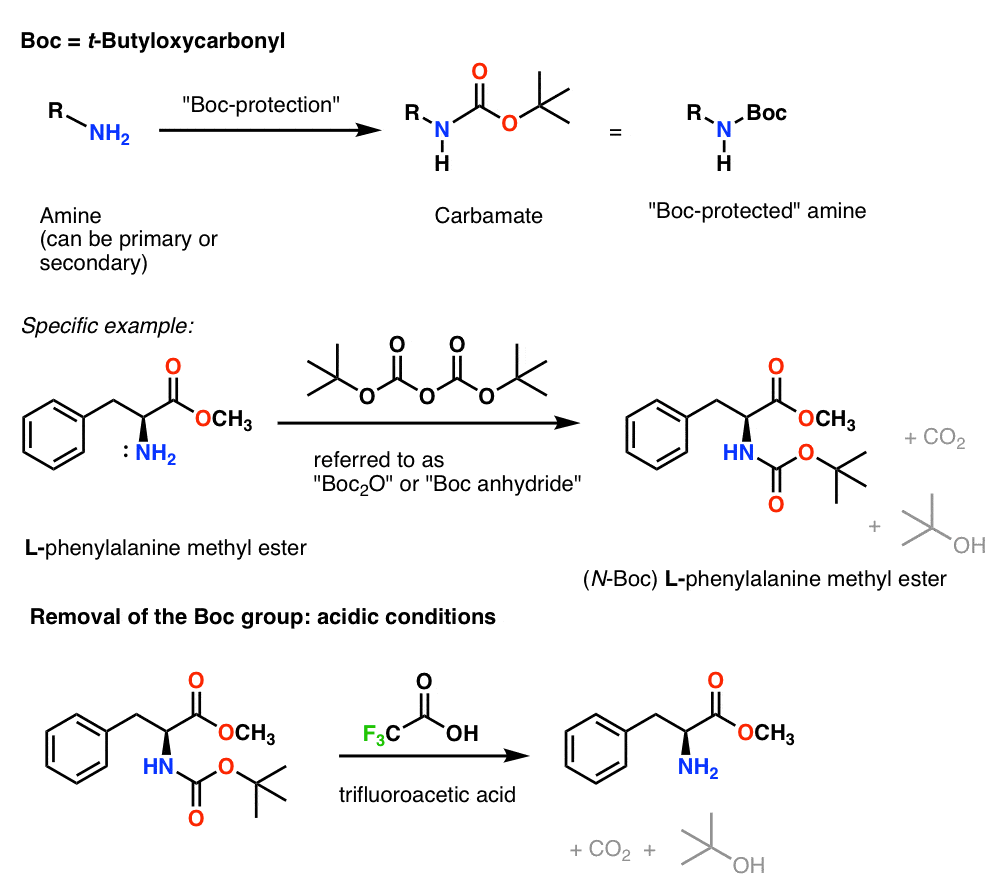
Instalacja i usuwanie grupy ochronnej „Boc”
Grupa Boc jest zwykle instalowana za pomocą „Boc2O” (czasami określana jako „bezwodnik Boc”), i usuwana za pomocą kwasu. Zazwyczaj wybiera się „czysty” (tj. nierozcieńczony) kwas trifluorooctowy (TFA), który usuwa grupy Boc bardzo czysto, uwalniając CO2 i alkohol t-butylowy.
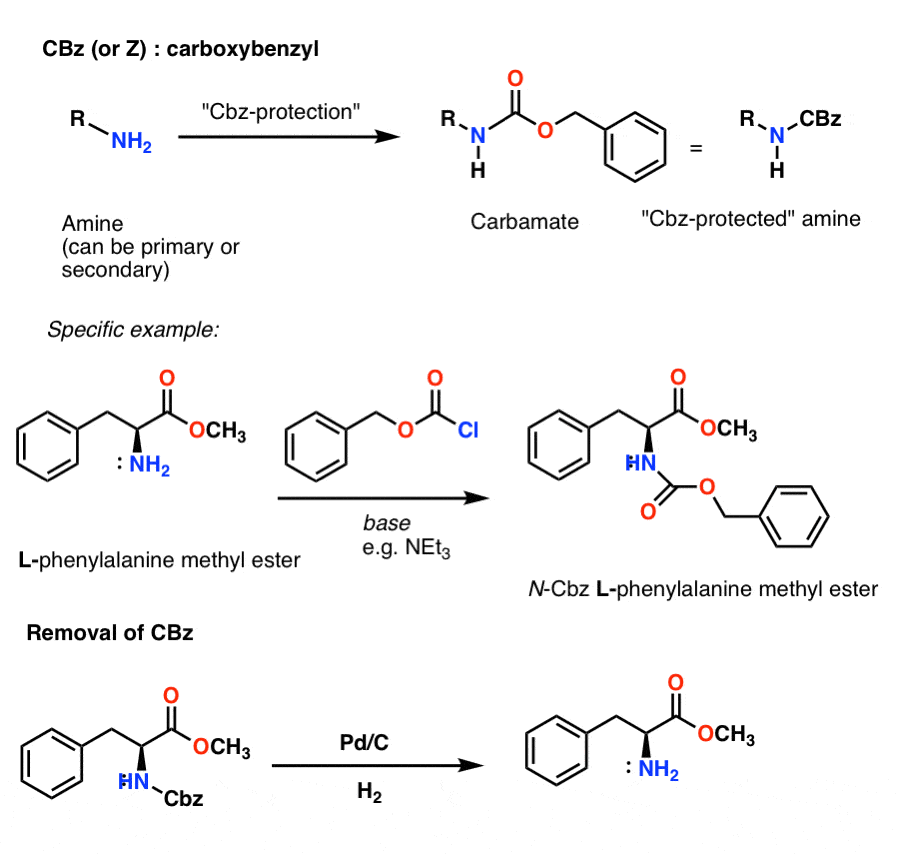
Instalacja i usuwanie karbaminianowej grupy ochronnej CBz (lub „Z”)
Grupę Cbz (czasami dalej oznaczaną skrótem „Z”) można zainstalować za pomocą CbzCl i łagodnej zasady, a zwykle usuwa się ją przez katalityczne uwodornienie (Pd-C/H2). Jest to bardzo łagodne i ma tę zaletę, że zachodzi przy neutralnym pH, pozostawiając grupy funkcyjne wrażliwe na kwas lub zasadę w spokoju.
Prosta synteza peptydów z wykorzystaniem karbaminianowych grup ochronnych
Powróćmy do syntezy peptydów i zastosujmy tę strategię grup ochronnych do wytworzenia Gly-Ala.
Zaczynamy od aminokwasu takiego jak L-alanina. Poddając alaninę działaniu Boc2O, otrzymujemy L-alaninę chronioną N-Boc. Następnym krokiem jest utworzenie chlorku kwasu przez użycie SOCl2. Po utworzeniu, dodajemy naszą aminę (np. L-walinę) w obecności nadmiaru zasady, tworząc nasze kluczowe wiązanie amidowe. Ostatnim krokiem do otrzymania dipeptydu jest deprotekcja aminy zabezpieczonej Boc kwasem trifluorooctowym (TFA), i voila! mamy nasz dipeptyd.
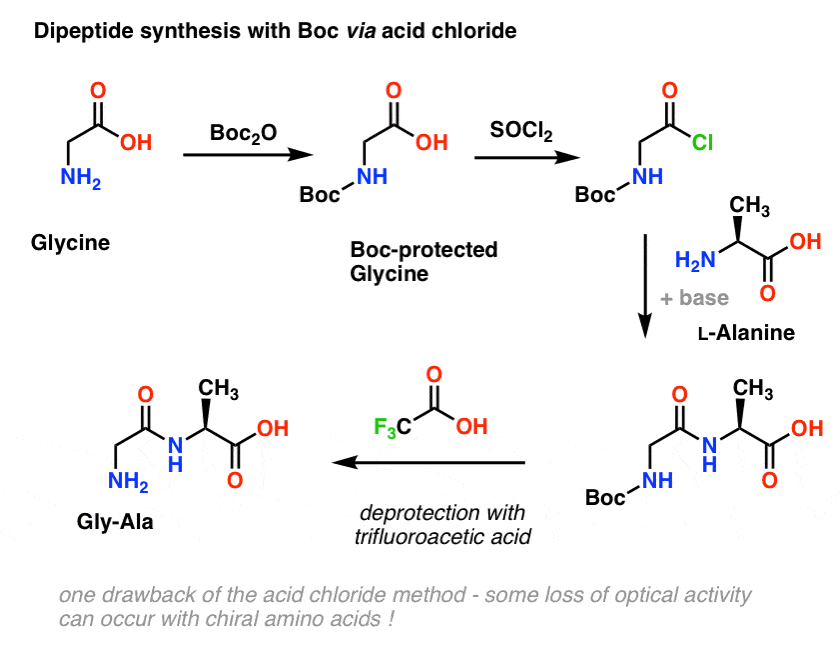
Mimo, że ta metoda może być dobra na papierze, jednym z problemów stosowania chlorków kwasu w praktyce jest to, że chiralne aminokwasy często tracą czystość optyczną przez tę metodę, proces czasami określany jako „racemizacja”, ale bardziej poprawnie nazywany jest „epimeryzacją” (technicznie bardziej poprawny, ponieważ wodór w centrum chiralnym jest odwrócony)
Ponieważ chiralność aminokwasów jest niezbędna dla ich funkcji biologicznej, zazwyczaj stosuje się nieco łagodniejszy protokół, który wykorzystuje DCC lub podobny odczynnik sprzęgający.
Tutaj, traktujemy glicynę chronioną Boc z DCC, aby aktywować kwas karboksylowy. Następnie dodajemy nasz nukleofil aminokwasowy (L-alaninę), który tworzy dipeptyd. Jeśli chcemy w tym momencie wyizolować dipeptyd Gly-Ala, możemy następnie usunąć grupę Boc za pomocą TFA.
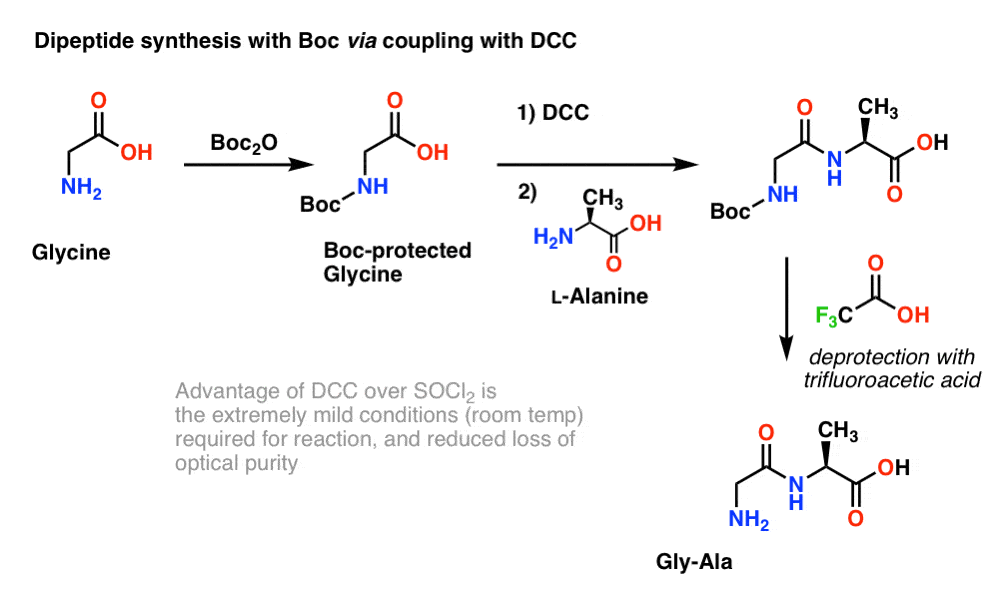
(uwaga o tym schemacie)
It Keeps Going…
Zauważ, że jeśli chcieliśmy utworzyć trójpeptyd, możemy po prostu kontynuować wykonywanie cykli dodawania DCC (aby aktywować kwas karboksylowy), a następnie dodawania nowych aminokwasów, budując peptyd po jednej jednostce na raz!
Istnieje szczególnie skuteczna metoda budowania dłuższych peptydów, której pionierem był Bruce Merrifield (i którą zastosowano między innymi w syntezie insuliny), zwana syntezą peptydów w fazie stałej, którą zajmiemy się następnym razem, gdy będziemy zajmować się tym tematem.
Przypisy
Uwaga 1. Podobnie jak w starym dowcipie o ekonomiście, który proponuje plan wydostania się z bezludnej wyspy:
„Załóżmy, że mamy łódź”.
Glicyna (jak wszystkie aminokwasy) sama jest zwitterionem. Traktowanie glicyny z SOCl2 powinien dać chlorek kwasu z protonowanej aminy. Powinien on być względnie stabilny w roztworze tak długo, jak długo nie zostanie dodana zasada.
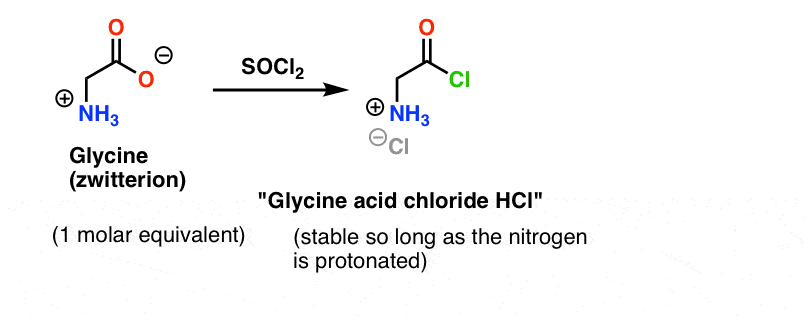
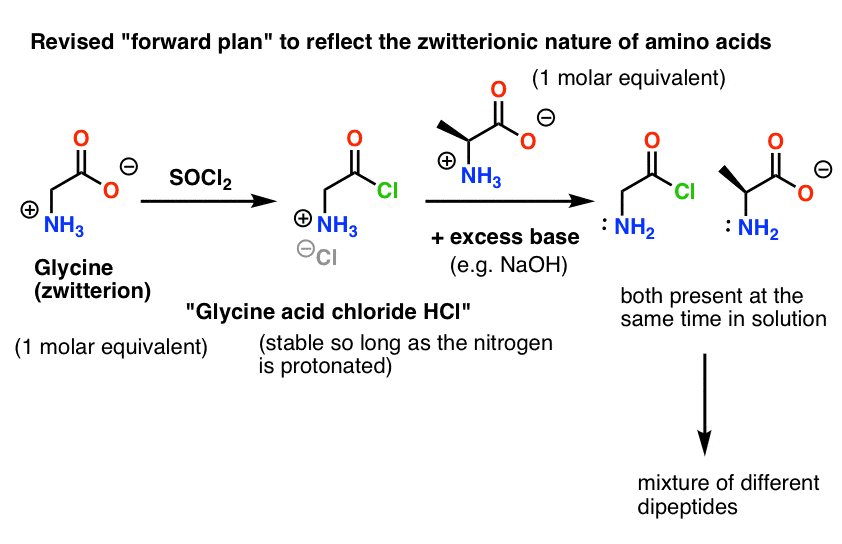
Tutaj jest problem. Ponieważ nukleofil naszego aminokwasu (alanina) jest również zwitterionowy, reakcja nie może zajść, dopóki nie dodamy nadmiaru zasady, aby uwolnić parę samotną na azocie alaniny. Po dodaniu zasady mamy w roztworze „chlorek kwasu glicynowego” i alaninę. Nie ma znaczącej różnicy w nukleofilowości między nitrogenami tych dwóch gatunków i każdy z nich będzie konkurował o reakcję z nukleofilem chlorku kwasu, co prowadzi do powstania mieszaniny Gly-Ala i Gly-GlyCl, a Gly-GlyCl może następnie reagować dalej z różnymi nukleofilami ustawionymi w roztworze, dając tri-, tetra- i wyższe peptydy.
Uwaga 2. John Sheehan, którego poznaliśmy wcześniej jako wynalazcę DCC w drodze do pierwszej syntezy penicyliny, również wykonał Gly-Gly chroniony ftalem poprzez syntezę Gabriela:
Referencja tutaj (JACS, 1949, 71, 1856)
Uwaga 3. Innym problemem związanym z użyciem amidowych grup ochronnych jest tworzenie azlaktonu, co może prowadzić do epimeryzacji chiralnych aminokwasów. Zobacz również ten zestaw problemów.
Uwaga 4. „Orthogonal Protecting Groups”. W planowaniu syntezy często niezwykle ważne jest posiadanie grup ochronnych, które są usuwalne w różnych warunkach. Właściwość ta jest często określana jako „ortogonalność”
Na przykład w poniższym dipeptydzie mamy dwie różne grupy ochronne na azocie – jedną Boc i jedną CBz. Wybierając „ortogonalne” grupy ochronne, każdy azot jest adresowalny – możemy wybrać, którą grupę ochronną usunąć, a nasza synteza może być kontynuowana od tego miejsca. Pozwala to uniknąć sytuacji, w której mamy dwie niezabezpieczone aminy i musimy polegać na tym, że jedna z nich jest bardziej reaktywna od drugiej. Takie podejście bardzo rzadko działa!
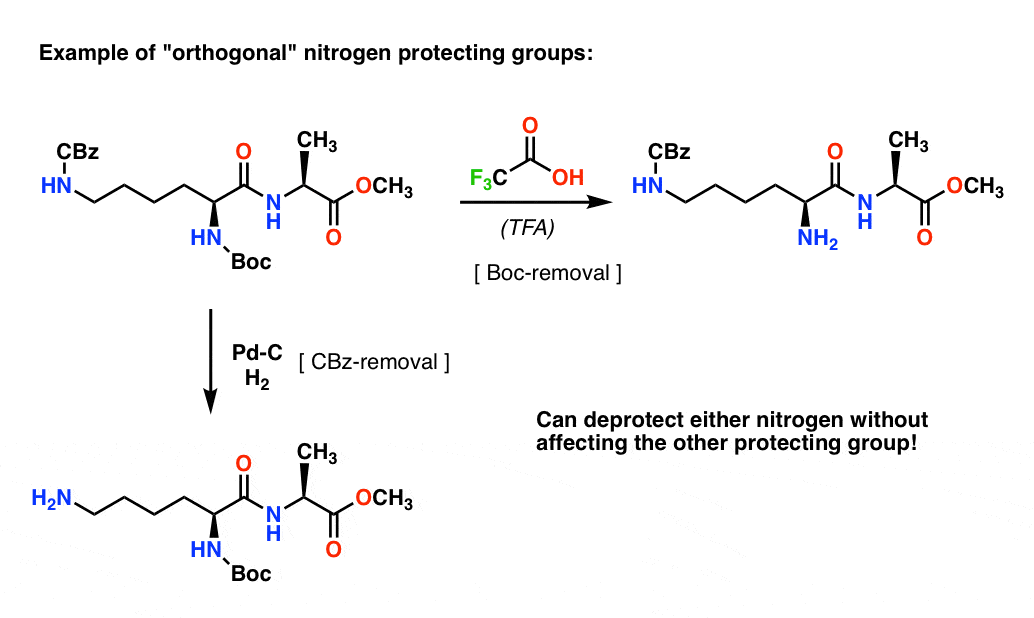
Jedna uwaga – dla uproszczenia alanina jest tu przedstawiona z wolnym kwasem karboksylowym, ale nieco lepszym podejściem byłoby użycie estru metylowego alaniny, aby uniknąć samosprzęgania między wolną aminą alaniny i wolnym kwasem karboksylowym.
Mate…
(Advanced) References and Further Reading
Karbaminiany są użyteczne jako grupy ochronne dla amin, a najczęściej stosowane to -Boc, -Cbz, i -Fmoc.
- Über ein allgemeines Verfahren der Peptid-Synthese
Max Bergmann and Leonidas Zervas
Ber. 1932, 65 (7), 1192-1201
DOI: 10.1002/cber.19320650722
Grupa ochronna -Cbz (karboksybenzylowa) została po raz pierwszy użyta przez Maxa Bergmanna i Leonidasa Zervasa w 1932 roku do syntezy peptydów i jest czasami oznaczana skrótem „-Z” na cześć Zervasa. - REMOVAL OF t-BUTYL AND t-BUTOXYCARBONYL PROTECTING GROUPS WITH TRIFLUOROACETIC ACID
Mechanisms, Biproduct Formation and Evaluation of Scavengers
Behrend F. Lundt, Nils L. Johansen, Aage Vølund, and Jan Markussen
J. Pept. Prot. Res. 1978, 12 (5), 258-268
DOI: 10.1111/j.1399-3011.1978.tb02896.x
W praktyce, nukleofilowe wymiatacze (np. tiole) są zwykle dodawane do koktajlu kwasowego (TFA) podczas deprotekcji, ponieważ deprotekcja Boc da elektrofilowe formy t-butylu (np. trifluorooctan t-butylu), które mogą reagować z wrażliwymi resztami (np. Trp lub Cys). - A Method of Synthesis of Long Peptide Chains Using a Synthesis of Oxytocin as an Example
Miklos Bodanszky and Vincent du Vigneaud
Journal of the American Chemical Society 1959, 81 (21), 5688-5691
DOI: 1021/ja01530a040
W pierwszej połowie XX wieku synteza peptydów była przeprowadzana przy użyciu standardowych technik chemii organicznej w fazie roztworu. Obecnie jest to znane jako LPPS (liquid-phase peptide synthesis). du Vigneaud otrzymał Nagrodę Nobla w dziedzinie chemii w 1955 roku za swoją pracę pokazującą, że synteza peptydów może być osiągnięta przy zastosowaniu właściwego doboru grup ochronnych i strategii syntetycznych. - TEMPERARY FORMATION OF THE AZLACTONE RING IN THE RACEMIZATION OF ACYL DERIVATIVES OF AMINO ACIDS WITH ACETIC ANHYDRIDE
Vincent du Vigneaud and Curtis E. Meyer
Biol. Chem. 1932, 99:143-151
http://www.jbc.org/content/99/1/143.citation
Innym problemem związanym z użyciem amidowych grup ochronnych jest powstawanie azylaktonu, co może prowadzić do epimeryzacji chiralnych aminokwasów. - A New Synthetic Route to Peptides
John C. Sheehan and Victor S. Frank
Journal of the American Chemical Society 1949, 71 (5), 1856-1861
DOI: 10.1021/ja01173a095
John Sheehan, wynalazca DCC w drodze do pierwszej syntezy penicyliny, również wytworzył Gly-Gly zabezpieczone ftalem poprzez syntezę Gabriela. - A New Method of Forming Peptide Bonds
John C. Sheehan and George P. Hess
Journal of the American Chemical Society 1955, 77 (4), 1067-1068
DOI: 1021/ja01609a099
Oryginalna praca na temat syntezy wiązań peptydowych/wiązań amidowych przy użyciu DCC. - Funkcja 9-fluorenylometoksykarbonylowa, nowa grupa aminoochronna wrażliwa na działanie zasad
Louis A. Carpino i Grace Y. Han
Journal of the American Chemical Society 1970, 92 (19), 5748-5749
DOI: 10.1021/ja00722a043 - 9-Fluorenylometoksykarbonylowa grupa aminoochronna
Louis A. Carpino i Grace Y. Han
The Journal of Organic Chemistry 1972 37 (22), 3404-3409
DOI: 10.1021/jo00795a005
Odkrycie i rozwój grupy ochronnej -Fmoc dla amin dodaje kolejną warstwę ortogonalności do strategii ochrony/deprotekcji amin. Grupa -Fmoc jest podatna na działanie zasad i w syntezie peptydów jest zwykle usuwana za pomocą 20% piperydyny w DMF. Cbz jest usuwana przez uwodornienie, -Boc usuwana kwasem, a -Fmoc zasadą.
.