W tym rozdziale przedstawione są takie zagadnienia, które dadzą studentowi pewien wgląd w metody analizy stosowane w badaniu technicznym żelaza i stali. Charakter tej pracy nie pozwala na szczegółowe traktowanie tematu – leczenie, które może wymagać oszacowania jednego lub więcej z następujących substancji: węgiel (wolny i połączony), siarka, krzem, fosfor, mangan, tytan, miedź, nikiel, kobalt, chrom, aluminium, arsen, antymon, cyna, wolfram, wanad, azot, żelazo. Ogólnie rzecz biorąc, szacunki najbardziej wymagane są te z węgla, siarki, krzemu i fosforu. Z innych pierwiastków i związków wymienionych, oznaczenia jednego lub więcej są wymagane w przypadku stali specjalnych. W celu uzyskania informacji dotyczących tych oznaczeń student jest odsyłany do Chemical Analysis and Foundry Chemistry, autorstwa Crobaugh ; The. Chemical Analysis of Iron, przez Blair ; „Carbon in Steel by Direct Combustion”, przez Blount, w The Analyst, Jan. 1902; „Sulphur in Wrought Iron and Steel”, przez Auchy, w Jour. Amer. Chem. Soc., marzec 1901, oraz inne artykuły w tych samych czasopismach. Student, który chce iść dalej powinien, jeśli to możliwe, uzyskać dostęp do prac i artykułów Campbella, Drown, i innych, publikowanych od czasu do czasu w różnych czasopismach chemicznych i metalurgicznych.
Jako że czas ucznia jest ograniczony, może on na razie odłożyć szacunki krzemu i fosforu, choć są one podane ze względu na ich znaczenie zarówno dla metalurga i odlewnika.
Aby uczeń mógł uzyskać bardziej dogłębne zrozumienie tematu, kilka uwag 0n składu i właściwości substancji rozważanych nie będzie nie na miejscu. Jeśli chodzi o wpływ różnych pierwiastków na stal, należy zapoznać się z The Manufacture and Properties of Structural Steel, autorstwa H. H. Campbella.
Węgiel występuje w żelazie w trzech stanach – grafitowym, rozpuszczonym i połączonym. Oprócz tych, inne formy zostały zidentyfikowane przez mikroskop.
Siarka istnieje w żelazie głównie jako siarczek FeS. który jest rozpuszczalny w stopionym żelazie.
Fosfor istnieje jako fosforek żelaza, który jest całkowicie rozpuszczalny w stopionym żelazie.
Krzem tworzy krzemku żelaza, który również jest rozpuszczalny w stopionym żelazie.
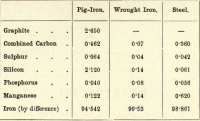
Z tych czterech elementów, a następnie, węgiel jest jedynym, który może istnieć w stanie wolnym. Różnice w proporcjach różnych elementów obecnych są prawie nieskończone, ale następujące krótkie zestawienie daje przybliżony skład surówki, kutego żelaza i stali, choć każdy z nich jest przedmiotem znacznej zmienności.
Uczeń jest zobowiązany do oszacowania następujących elementów:-
(1) Węgiel,
(a) Całkowity.
(b) Grafitowy.
(c) Połączony.
(2) Siarka.
(3) Krzem. (Jeśli czas na to pozwala.)
(4) Fosfor. (Jeśli czas pozwoli.)
WĘGIEL Razem
W tym szacunku węgiel jest przekształcany w CO2, który jest wchłaniany w żrącym potażu. Z masy CO2 w ten sposób uzyskane węgla jest obliczana.
Na pierwszy rzut oka wydaje się, że najprostszą procedurą byłoby zapalić żelaza lub stali otworów bezpośrednio w prąd tlenu i wchłonąć CO2 w ten sposób utworzony w KHO. Niestety ta metoda, jak dotąd, albo okazały się niedokładne, lub gdy całkowite spalanie zostało uzyskane, aparat niezbędny do wytrzymania wysokiej temperatury lub innych zmian w leczeniu nie był odpowiedni do pracy technicznej (patrz artykuły Blount w The Analyst). Student stwierdzi, że metoda tutaj podana nie jest w żaden sposób idealna, z technicznego punktu widzenia, na wynik wygody i szybkości, i wydaje się, że istnieje prawdopodobieństwo, że zostanie zastąpiona w najbliższej przyszłości przez niektóre szybsze metody bezpośredniego utleniania.
Metoda przyjęta.-Po odniesieniu do wielu prac na ten temat, duża różnorodność metod zostanie znaleziony. Metoda tutaj podana będzie ze zwykłą starannością dać dokładne wyniki. W skrócie wygląda to następująco:-
Żelazo rozpuszcza się w roztworze podwójnego chlorku potasu i miedzi, zakwaszonym HCl. Miedź metaliczna zostaje wytrącona i ponownie rozpuszczona; żelazo zostaje rozpuszczone, a węgiel pozostaje w zawiesinie. Następnie zbiera się go i zapala w piecu do spalania z tlenem, a wydzielający się CO2 waży się.
Roztwarzanie żelaza.- Odważyć 1 gram odłamków surówki. Przenieść do zlewki o pojemności 300 cm3. Dodać 100 cm3. CuCl2,2KCl,2H2O, który sporządza się w następujący sposób. Rozpuścić w wodzie 149,1 części KCl i 170,3 części skrystalizowanego CuCl2, 2H2O. Odparować i wykrystalizować podwójny chlorek. Rozpuścić 300 g soli podwójnej w wodzie destylowanej. Przefiltrować przez zapalony azbest i przechowywać w szklanych butelkach z korkiem.
Do zawartości zlewki dodać 7 c.cs. HCl, aby roztwór stał się kwaśny. Mieszać z przerwami, aż do rozpuszczenia żelaza. Umieścić zlewkę z zawartością pod koniec roztworu na łaźni wodnej w temperaturze około 60° C. Zachodzą następujące reakcje-Fe + CuCl2 = FeCl2 + Cu i Cu + CuCl2 = 2CuCl. KCl jedynie wspomaga rozpuszczanie wytrąconej miedzi. Po około 40 minutach od dodania podwójnych chlorków roztwór powinien być prawie pełny, a większość miedzi rozpuszczona. Spłukać ścianki zlewki odrobiną zakwaszonego podwójnego chlorku. Do roztworu dodać odrobinę zapalonego azbestu, aby osadzić substancje węglowe i zapobiec ich zatykaniu filtra (zgodnie z zaleceniami Barby).
Do filtracji bardzo wygodne są specjalne łódki platynowe, wyposażone na zasadzie tygla Goocha. Uczeń może jednak odfiltrować substancje węglowe za pomocą tygla Goocha, wspomagane przez sztuczne ssanie, substancja węglowa jest wypłukiwana strumieniem wody po cieczy przeszedł przez filtr. Ostrożnie przemyć węgiel na filtrze gorącą wodą. Wysuszyć tygiel i
zawartość w piecu powietrznym w temperaturze 100 C. Substancje węglowe są teraz gotowe do zapłonu.
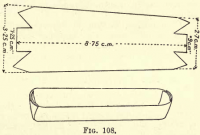
Utlenianie węgla.-Przygotować łódź platynową przez cięcie kawałka folii platynowej, jak na rys. 108, składając boki i końce, aby utworzyć koryto. Przeniesienie węgla i azbestu z Gooch do łodzi.
Piec do spalania, akcesoria i wyposażenie muszą być ustawione w porządku. Aparat do oczyszczania tlenu jest ponownie używany, ale tym razem jest wyposażony w trójdrożną rurę, z kranami umieszczonymi pomiędzy magazynem a oczyszczaczami. Pozwala to na przepływ powietrza przez aparat. Rura spalania może być wykonana z twardego szkła jenajskiego, porcelany lub platyny. Pomiędzy piecem a cebulkami potażu stosuje się dwie rury U. Końcówka pierwszej rurki znajdująca się najbliżej pieca zawiera bezwodny CuSO4, a druga końcówka bezwodny CuCl. Druga rura U zawiera suszony CaCl2. Te dwie rurki tworzą „ciąg oczyszczający”. CuCl pochłania każdy Cl, a pozostałe substancje każde H2O. Zestaw ten będzie służył do wielu oznaczeń. Po nim następują bańki potasowe i rurki ochronne, a aspirator powinien być pod ręką, aby w razie potrzeby móc zaczerpnąć prąd powietrza przez aparat. Bańki potażu są ładowane z 8E. KHO, a rurka ochronna z CaCl2. Test pieca i żarówki, jak opisano wcześniej (patrz Węgiel i Koks), rura jest ładowana jak na szkicu, łódź jest obecnie przechowywane w piecu powietrza w 100 ° C.
Gdy wszystko jest gotowe, palniki zostały wyłączone na jakiś czas, łódź i zawartość są wstawiane. Palniki są zapalane od przodu, stopniowo pracują do tyłu, a powolny prąd tlenu, około dwóch pęcherzyków na sekundę, po wcześniejszym włączeniu, aż rura jest pełna tlenu. Regulować temperaturę, aż łódź jest w matowej czerwieni, a jeśli roztwór w żarówki wykazuje oznaki uruchomienia z powrotem do pieca, zwiększyć prąd tlenu do trzech lub czterech pęcherzyków na sekundę.
Od czasu umieszczenia w łodzi około pięćdziesięciu minut wystarczy do całkowitego spalania. Wyłącz tlen i przepuszczaj prąd powietrza przez dziesięć minut.
Bańki potasowe i rurka ochronna są teraz usuwane i ważone, a węgiel obliczany jak zwykle.
(b) Węgiel grafitowy -Żelazo jest przez niektórych rozpuszczane w HCl, przez innych w HNO3, gdy węgiel grafitowy pozostaje jako pozostałość. Dla surówki albo metoda, z ostrożnością, daje dobre wyniki, ale dla stali zawierających grafit Blair zaleca roztwór w kwasie azotowym. (Dla tej metody skonsultować się z Blair.)
Odważyć 5 gms. odwiertów surówki. Rozpuścić w 50 c.cs. SE. HCl przy pomocy delikatnego ciepła. Gotować przez kilka minut. Rozcieńczyć do 100 c.cs. (prawie). Przefiltrować przez tygiel Goocha. Przemyć gorącą wodą, a następnie wrzącą E. KHO. (To rozpuszcza wszelkie SiO2.) Przemyć ponownie gorącą wodą, aby usunąć KHO. Wysuszyć tygiel i zawartość.
Oszacować węgiel jak poprzednio przez spalanie, i obliczyć procent jak zwykle.
(c) Combined węgla (przez różnicę).-Całkowity węgiel i węgiel grafitowy jest znany, łączny węgiel uzyskuje się przez odjęcie grafitowego od węgla całkowitego.
W sprawie bezpośrednich metod szacowania należy skonsultować się z wymienionymi organami.
Oszacowanie zawartości siarki w żelazie i stali
Istnieją znaczne różnice w opiniach na temat najlepszej metody szacowania zawartości siarki w żelazie i stali. Stara metoda roztworu wodnego i strącania BaCl2 jest uznawana za bardzo niedokładną, ale powolne roztwarzanie w HNO3, z bardzo małą ilością lub bez HCl, a następnie ostrożne strącanie BaCl2 w obecności zdecydowanego nadmiaru HCl i z należytą starannością co do czasu i warunków strącania oraz środków ostrożności przeciwko zanieczyszczeniu osadu żelazem – z tymi i starannością można uzyskać dobre wyniki. Blair, z drugiej strony, zaleca roztwór w HCl, S jest wydzielany jako H2S, który jest wchłaniany do roztworu (alkaliczne) Pb(NO3)2 tworząc PbS, który jest rozpuszczony w HCl + KClO3, a S wytrąca się jako BaSO4. Dalsze metody patrz Blair, Stillman, Auchy, Crobaugh i Drown. Inną metodą w powszechnym użyciu jest ewolucja S jako H2S, a następnie absorpcja w roztworze chlorku kadmu. Wytrącony siarczek kadmu rozpuszcza się w HCl i S oszacowane przez miareczkowanie roztworem jodu, lub bardziej powszechne jeszcze, H2S jest absorbowany w wodzie Br., a następnie wytrąca się jako BaSO4 lub jest absorbowany w NaOH i miareczkowany jodem; ten ostatni jest ulubioną metodą. (Patrz Blair.) Następująca metoda jest tutaj podana:-
Outlenianie przez HNO3 (tak zwana metoda Aqua Regia).- Odważyć 5 gms. odłamków i przenieść do głębokiej zlewki o pojemności 200 cm3 . Ostrożnie dodać około 40 cm3. 16E. HNO3, partiami po ok. 10 cs. jednorazowo, przykrywając zlewkę dużym szkiełkiem zegarkowym i uważając, aby działanie nie było zbyt gwałtowne. Po ustaniu działania należy sprawdzić, czy wszystkie cząsteczki są rozpuszczone (z wyjątkiem węgla). Jeśli nie, podgrzać na łaźni piaskowej, dodać 3 lub 4 krople 16E. HCl, i ogrzewać do rozpuszczenia.
Gdy roztwór jest kompletny, dodać trochę Na2CO3, aby przekształcić H2SO4 w Na2SO4, który jest nielotny przy odparowywaniu.
Usuń z łaźni piaskowej, i dodać 5 c.c. silnego HCl w nadmiarze koniecznym do rozpuszczenia związków żelaza wytrącanych przez Na2CO3. Odfiltrować SiO2 i C. Dobrze przemyć. Odparować do sucha, aby SiO2 stał się nierozpuszczalny. Uzupełnić HCl i odparować, aż Fe2Cl6 zacznie się krystalizować. Następnie dodać 5 c.cs. HCl. i przefiltrować w przypadku obecności pozostałości. (Jeśli nie ma żadnych pozostałości, SiO2 nie był w roztworze i można było pominąć odparowanie). Przefiltrować i ostrożnie przemyć osad w Gooch, doprowadzając ciecz i popłuczyny do około 100 c.cs.
Ogrzać do wrzenia. Dodać 10 cs. nasyconego roztworu BaCl2. Gotować przez 30 minut. Pozostawić na noc. Przefiltrować przez filtr Goocha. Przemyć niewielką ilością E. HCl. a następnie wodą. Wysuszyć, zapalić i zważyć jak zwykle BaSO4, które powinny być białe, a nie zanieczyszczone solami żelaza.
Obliczyć procent S w zwykły sposób. Ponieważ niektóre z używanych odczynników mogą zawierać siarkę, należy przeprowadzić ślepą próbę, stosując takie same ilości jak w rzeczywistej analizie, a znalezioną siarkę odjąć od poprzedniego wyniku.
WYKRYWANIE SILIKONU
Podana tu metoda jest metodą Drowna, jest szybka i dokładna. Żelazo rozpuszcza się w HNO3, a następnie w H2SO4, odparowując do sucha. Po tym następuje rozwiązanie, pozostawiając krzem w pozostałościach jako SiO2.
Szczegóły.- Odważyć 2 gms zwojów i przenieść do platynowego lub porcelanowego naczynia. Dodać 30 c.cs. 8E. HNO3 Gdy działanie pozornie ustanie, dodać 20 cs. 18E. H2SO4, i odparować. (Blair zaleca delikatny podmuch gorącego powietrza igrający na powierzchni cieczy. Powietrze jest ogrzewane przez przepuszczanie go przez małą spiralę z miedzianej rury ogrzewanej nad bunsenem. Parowanie jest w ten sposób przyspieszone i zapobiega spirali). Kontynuować parowanie aż do pojawienia się obfitych oparów SO3. Ostudzić i ostrożnie rozcieńczyć wodą destylowaną do 130 cm3 . Podgrzewać do rozpuszczenia FeSO4. Przefiltrować i przemyć najpierw niewielką ilością E. HCl, a następnie gorącą wodą. Filtracja ta jest najlepiej wykonywana przy użyciu 7 cm. bezpopiołowej bibuły filtracyjnej (sprawdzić popiół przez zapalenie dwóch lub trzech bibuł). Wysuszyć, przenieść do tygla platynowego, zapalić jak zwykle i zważyć. Do tygla dodać 5 cs. silnego H2SO4 i 5 cs. silnego HF. Ostrożnie odparować do sucha, stosując gorący podmuch powietrza, aby przyspieszyć odparowanie. Zapalić i ponownie zważyć. Zakładając, że H2SO4 i HF są czyste, różnica w masie stanowi SiO2. Sprawdzić zawartość H2SO4 i HF (zwłaszcza tego ostatniego) przez odparowanie ślepej próby. Każda znaleziona pozostałość musi być dopuszczalna.
WYKRYWANIE FOSFORU
Tutaj, ponownie, liczne metody są podane przez różne władze, większość z nich daje dokładne wyniki, gdy są starannie przestrzegane. Dwie metody najbardziej odpowiednie do analizy technicznej to metoda redukcji objętościowej przygotowana przez podkomitet (panowie Barba, Blair, Drown, Dudley i Shimer) Międzynarodowego Komitetu Standardów Stali, U.S.A., oraz zmodyfikowana metoda redukcji podana przez panów Dudley i Pease, Jour. Anal. Appl. Chem., vii. 108. Pierwsza metoda jest w pełni omówione w Blair’s Analysis of Iron; druga metoda jest podana tutaj.
Żelazo rozpuszcza się, a P wytrąca się jako fosfomolibdenian amonu. To jest rozpuszczony, a przez działanie Zn i H2SO4 MoO3 jest zmniejszona, a zredukowana ciecz jest następnie miareczkowany z K2Mn2O8 (roztwór wzorcowy), a od liczby c.c. używane zawartości P może być obliczona.
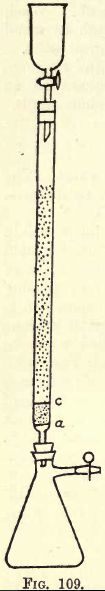
Szczegóły. -Gdzie trzeba wykonać dużo pracy, konieczny jest aparat do wstrząsania (patrz Katalogi Dostaw Chemicznych). Uczeń może jednak wykonać niezbędne wytrząsanie ręcznie. Przed przystąpieniem do analizy, aparat redukcyjny (modyfikacja reduktora Jonesa) musi być przygotowany (patrz rys. 109).
Na a jest drobno perforowany dysk z mocnej folii platynowej. Pomiędzy a i c jest około ¾ cala czystego białego piasku, c jest kolejnym perforowanym dyskiem platynowym.
Powyżej tego dysku rura jest wypełniona drobnym granulatem amalgamatu cynku, przygotowanym w następujący sposób:- Rozpuścić 5 gms. Hg w 25 cm3 silnego HNO3, rozcieńczając wodą i doprowadzając roztwór do objętości 1 litra. Do tego roztworu wsypać pół kilograma granulowanego cynku, który przejdzie przez sito 20, ale nie 30. Wstrząsać przez jedną lub dwie minuty. Odlać roztwór. Umyć i wysuszyć cynk, który jest teraz amalgamowany. Lejek i kolba są przymocowane do aparatu w sposób przedstawiony na rysunku.
Przygotować następujące odczynniki :-
(a) Silnie utleniający roztwór K2Mn2O8. 12 g czystego K2Mn2O8 w 1 litrze wody. Filtr i butelka.
(b) Roztwór molibdenianu.- Rozpuścić 50 gms. MoO3 w 200 c.cs, NH4HO (S.G. .96). Przefiltrować, a do przesączu dodać 500 c.cs. HNO3 (S.G. 1.2). Pozostawić na co najmniej 24 godziny przed użyciem.
(c) Roztwór kwaśnego siarczanu amnwitu.- Do 500 cm3 wody destylowanej dodać 27,5 cm3. NH4HO (S.G. 0,96), a następnie 24 cs. czystego H2SO4 (S.G. 1,84) i rozcieńczyć do 1000 cs.
(d) Roztwór wzorcowy K2Mn2O8.- Rozpuścić 2 gms. skrystalizowanego K2Mn2O8 w 1000 cs. wody destylowanej. Roztwór standaryzować w następujący sposób: Odważyć 3 partie od .1 do .3 gm. każdy z dokładnie oczyszczonego drutu żelaznego, którego zawartość żelaza jest znana. Przenieść do kolby Erlenmeyera o pojemności 100 cm3 i dodać do każdej z nich po 40 cm3. 8E. H2SO4. Po rozpuszczeniu gotować 5 minut, rozcieńczyć do 150 ml, przepuścić przez reduktor i przemyć, doprowadzając objętość do 200 ml, zgodnie z zaleceniami analizy. Miareczkować każdą partię za pomocą K2Mn2O8. Wyniki powinny być zgodne dla żelaza metalicznego z dokładnością do 1/100 miligrama. Dokonać wymaganych poprawek na zanieczyszczenia w pobranym drucie. Załóżmy, że 1 c.c. K2Mn2O8 = .0034923 gm, Fe, następnie pomnożyć tę wartość w Fe przez stosunek MoO3 do Fe, a mianowicie, .9076, a produkt przez stosunek P
obecny do MoO3, a mianowicie, .019, mamy
1 c.c. K2Mn2O8 = .0000602 gm. P
Analiza
Odważyć odwierty o masie 1 gm. Przenieść do kolby Erlenmeyera o pojemności 200 cm3. Dodać 70 cs. 5E. HNO3. Gdy roztwór jest pełny, gotować przez minutę i dodać 10 cm3 „utleniającego” roztworu K2Mn2O8. Gotować do zaniku różowego zabarwienia i wydzielenia się MnO2. Wyjąć i dodawać stopniowo, mieszając, kryształki czystego (nie zawierającego fosforu) FeSO4 aż do sklarowania zawartości. Ogrzać roztwór do temp. 80°C (jeśli jest obecny As, do temp. 35°C). Dodać 75 c.c. roztworu molibdenianu w temperaturze 27° C. Kolbę zamknąć korkiem gumowym i wstrząsać przez 5 minut. Odstawić na 5 minut. Następnie przesączyć przez 9 cm, filtr, i przemyć kwaśnym roztworem siarczanu amonu, aż kilka kropli przemywających daje żadnego koloru z siarczkiem amonu.
Rozpuścić osad na papierze z 5 c.cs. NH4HO (S.G. 0,90) i 25 cs. wody, pozwalając roztworowi spłynąć z powrotem do pierwotnej kolby, co spowoduje rozpuszczenie osadu przylegającego do jej ścianek. Płukać, aż przesącz i popłuczyny wyniosą 150 cm3. Dodać 10 cm3 mocnego H2SO4 (S.G. 1,84) i rozcieńczyć do 200 cm3. Roztwór jest teraz gotowy do redukcji.
Wlać 100 c.c. ciepłego ~E/2 H2SO4 do lejka. Podłączyć kolbę do pompy filtracyjnej i otworzyć zacisk, tak aby roztwór prawie, ale nie całkiem, wypłynął z lejka. Następnie do lejka dodać następującą ślepą próbę 5 c.c. NH4HO (S.G. 0,90), 10 cs. H2SO4 (S.G. 1.84), i 50 cs. wody, zmieszanych razem. Ponownie otworzyć zacisk, tak aby prawie wypłynęła ta mieszanina z lejka. Teraz dodać 200 c.cs, E/2 H2SO4 do lejka, i prawie przez niego przepłynąć.
Odłączyć kolbę, najpierw zamykając kurek olejowy lejka. Miareczkować zawartość kolby z K2Mn2O8. Ogólnie około 0,1 c.c. nadmanganianu są w ten sposób zużyte, a ilość ta musi być odjęta od przyszłych odczytów.
Teraz przenieść roztwór do redukcji do lejka. Przymocować czystą kolbę. Podłączyć i uruchomić pompę filtrującą. Otworzyć kurek odcinający i zacisk, tak aby prawie przepuścić roztwór. Wypłukać kolbę, w której znajdował się roztwór, za pomocą 100 c.cs. E/2 H2SO4. Dodać to do lejka i postępować jak poprzednio.
Na koniec dodać i prawie przepuścić kolejne 100 c.c. kwasu.
Zredukowany roztwór w kolbie filtracyjnej powinien być teraz jasnozielony.
Usunąć jak poprzednio i miareczkować roztworem nadmanganianu. Zieleń zmienia się na różowobrązową, następnie różowawożółtą, potem bezbarwną, a w końcu uzyskuje się trwały róż (po odstaniu jednej minuty). Od uzyskanego odczytu odjąć odczyt ślepej próby i obliczyć procent P obecny na podstawie danych podanych powyżej.
Zamiast tej metody objętościowej, niektórzy chemicy wolą ważyć bezpośrednio żółty osad fosfo-molibdenianu. Szczegóły patrz Blair’s Analysis of Iron, str. 108.
Uwaga.-Student powinien, gdziekolwiek to możliwe, korzystać z odniesień do specjalnych organów. Do tego czasu powinien być w stanie konsultować, porównywać i, do pewnego stopnia, rozsądnie korzystać z takich materiałów. Żaden podręcznik nie może dać w każdym razie kompleksowe traktowanie „Żelaza i stali,” lub, dla tej sprawy, z jednego z tematów traktowanych w tej sekcji, dlatego takie odniesienia, jak podano, wraz z bieżącą literaturą, musi być starannie przeglądane przez analityka, który chce doskonalić się w pracy technicznej. Podano kolorymetryczne oznaczanie węgla związanego metodą Eggertza; mangan może być oznaczany podobnie metodą kolorymetryczną Petera lub metodą octanową (patrz Blair, itp.).
.